Reparación do ADN Índice Danos no ADN | Mecanismos de reparación do ADN | Resposta global aos danos no ADN | Reparación do ADN e envellecemento | Medicina e modulación da reparación do ADN | Reparación do ADN e cancro | Reparación do ADN e evolución | Notas | Véxase tamén | Menú de navegación5055816510672810.1111/j.1532-5415.1971.tb02577.x"DNA damages"1558949010.1016/j.amjmed.2004.06.033"Nobel Prize in Chemistry Awarded to Tomas Lindahl, Paul Modrich and Aziz Sancar for DNA Studies""THE NOBEL PRIZE IN CHEMISTRY 2015 - DNA repair – providing chemical stability for life""Association of hOGG1 Ser326Cys polymorphism with susceptibility to hepatocellular carcinoma"4537974"Tumour suppressor gene"o orixinal"DNA damage tumor suppressor genes and genomic instability"1510879910.1016/j.gde.2003.12.003"DNA damage by reactive species: Mechanisms, mutation and repair"22750987"DNA Adducts: Endogenous and Induced""DNA Packaging: Nucleosomes and Chromatin""Histone modifications in response to DNA damage""DNA damage and mutation. Types of DNA damage."10.7750/BioDiscovery.2014.11.1"Sources of DNA Damage""The DNA-damage response in human biology and disease"290670010.1038/nature08467"Cancer Biologists Find DNA-Damaging Toxins in Common Plant-Based Foods. Liquid smoke, black and green teas and coffee produced levels of cell DNA damage comparable to chemo drugs.""Chemical-induced DNA Damage and Human Cancer Risk"1054770210.1146/annurev.micro.53.1.5770340-67171128145610.1007/s004390000447"Endogenous DNA damage in humans: a review of quantitative data."10.1093/mutage/geh025"Oxidative DNA damage: mechanisms, mutation, and disease"10.1096/fj.02-0752revdoi 10.1016/S0065-230X(08)60702-2Damage to DNA Caused by Hydrolysis (volume 40 of the series NATO Advanced Study Institutes Series)DNA Damage RecognitionEffects of Solar Ultraviolet Photons on Mammalian Cell DNA1701516710.1016/j.freeradbiomed.2006.06.011306582610.3123/jemsge.28.56"DNA Damage: Chemistry, Repair, and Mutagenic Potential"10.1006/rtph.1996.0002"Hydrogen peroxide induces DNA single- and double-strand breaks in thyroid cells and is therefore a potential mutagen for this organ."1950906510.1677/ERC-09-0020"Polycyclic Aromatic Hydrocarbon-DNA Adducts in Liver Tissues of Hepatocellular Carcinoma Patients and Controls"11948486"Carcinogenic polycyclic aromatic hydrocarbon-DNA adducts and mechanism of action"1098-228010.1002/em.20095"Eukaryotic chromosome"26309809"Oxidative stress induces degradation of mitochondrial DNA."267786710.1093/nar/gkp100"ROS-Generating Mitochondrial DNA Mutations Can Regulate Tumor Cell Metastasis"1654063110.1158/0008-5472.CAN-05-40061646023010.1089/dna.2006.25.691766795410.1038/nrm2233"DNA damage""Mutation""Nuclear DNA damage as a direct cause of aging"1959432810.1089/rej.2009.0847The Cell: A Molecular ApproachDNA Repair Pathways and Mechanisms1279782910.1021/cr0204348"Evolution of Mutation Rates: Phylogenomic Analysis of the Photolyase/Cryptochrome Family"26688311922892210.1093/molbev/msp029327889810.1002/em.28501102101679337010.1016/S0076-6879(06)08012-8924241110.1038/41365"Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae"2312048628283"Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways"45224988901831043854210.1074/jbc.274.33.23599"Processing of DNA for nonhomologous end-joining by cell-free extract"5496221569256510.1038/sj.emboj.7600563"Biochemical evidence for Ku-independent backup pathways of NHEJ"2033131295477410.1093/nar/gkg7281474443910.1016/S0092-8674(04)00039-X"Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells"36515032361043910.1073/pnas.1213431110"Homology and enzymatic requirements of microhomology-dependent alternative end joining"43859362578997210.1038/cddis.2015.581601216710.1074/jbc.M503776200"Alternative end-joining mechanisms: a historical perspective"36136182356511910.3389/fgene.2013.00048"Repair of strand breaks by homologous recombination"38095762409790010.1101/cshperspect.a01274094035110.1016/s0022-5193(76)80025-21139541210.1146/annurev.biochem.70.1.3691700645010.1038/nature05160"Eukaryotic Translesion Polymerases and Their Roles and Regulation in DNA Damage Tolerance"26508911925853510.1128/MMBR.00034-08"Translesion DNA Synthesis and Mutagenesis in Eukaryotes"10.1101/cshperspect.a012708"Translesion DNA synthesis in eukaryotes: A one- or two-polymerase affair"10.1101/gad.10098021861629410.1021/bi800529f2130294310.1021/bi102064z"Translesion DNA Synthesis"1142551210.1016/S0921-8777(01)00089-110.1046/j.1365-2184.2003.00266.x1255688410.1038/nature013681166571910.1016/s0065-230x(01)83007-4"The role of the cyclin-dependent kinase inhibitor p21 in apoptosis"12479224118337681788340810.1111/j.1574-6976.2007.00082.x1646401210.1021/cr04049511615317310.1146/annurev.micro.59.031805.133658"Shorter telomeres, accelerated ageing and increased lymphoma in DNA-PKcs-deficient mice"12990481510582510.1038/sj.embor.74001271195099810.1126/science.10701741647282710.1016/j.mrfmmm.2005.11.008158583571592723510.1016/j.mad.2005.03.0161124208510.1038/350656381520547710.1126/science.10991961254739210.1016/S1568-7864(02)00219-71473463510.1096/fj.03-0890fje1083094810.1038/350126931698787810.1093/hmg/ddl214"Nucleotide excision repair and human syndromes"10.1093/carcin/21.3.4532511965810.1007/s00249-014-0981-x16849332"The premature ageing syndromes:insights into the ageing process"2476845110.1016/j.dnarep.2014.03.019"Nucleotide excision repair deficiency is intrinsic in sporadic stage I breast cancer"10.1073/pnas.09147721071711905510.1158/1055-9965.EPI-06-0449"Unearthing Prehistoric Tumors, and Debate"Molecular biology of the cell1617194310.1016/j.canlet.2005.08.003"Deficiency of human BRCA2 leads to impaired homologous recombination but maintains normal nonhomologous end joining"10.1073/pnas.15125349810.1002/0470025077.chap84b1649107010.1038/nrc1799"Epigenetic modifications in cancer"35908022208234810.1111/j.1399-0004.2011.01809.x"Negative regulation of BRCA1 gene expression by HMGA1 proteins accounts for the reduced BRCA1 protein levels in sporadic breast carcinoma"1507341264010910.1128/MCB.23.7.2225-2238.20031741271210.1093/mutage/gem009"Epigenetic changes of DNA repair genes in cancer"30309732127845210.1093/jmcb/mjq053"Epigenetic field defects in progression to cancer"36486622367173010.4251/wjgo.v5.i3.43New Research Directions in DNA Repair. Chapter 16: DNA Damage, DNA Repair and Cancer"Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2"20332909635610.1073/pnas.94.7.3122"Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6"26129361672843310.1093/carcin/bgl079"Disruption of Brca2 increases the spontaneous mutation rate in vivo: synergism with ionizing radiation"10840101185039710.1093/embo-reports/kvf037"Bloom's syndrome. I. Genetical and clinical observations in the first twenty-seven patients"17064305770175"Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island"24917231870415910.1371/journal.pgen.1000155"DNA damage, homology-directed repair, and DNA methylation"19131001761697810.1371/journal.pgen.0030110"Li-fraumeni syndrome"31356492177951510.1177/1947601911413466935317710.1126/science.278.5340.1043"O(6)-methylguanine methyltransferase in colorectal cancers: detection of mutations, loss of expression, and weak association with G:C>A:T transitions"17745511588878710.1136/gut.2004.0595351617485410.1093/jnci/dji275"Promoter methylation status of hMLH1, MGMT, and CDKN2A/p16 in colorectal adenomas"29095552065306410.3748/wjg.v16.i28.35532170623310.1007/s00423-011-0812-92342209410.1158/1078-0432.CCR-12-35182327113310.1007/s11033-012-2465-31588709910.1053/j.gastro.2005.01.056"Modulation of mismatch repair and genomic stability by miR-155"28724632035127710.1073/pnas.1002472107"Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer"33510282249482110.1186/2041-9414-3-3"Human DNA Repair Genes""MicroRNA-182-5p targets a network of genes involved in DNA repair"35430902324974910.1261/rna.034926.1122228676910.1038/onc.2011.660DNA Repair, Genetic Instability, and Cancer"The consequences of Rad51 overexpression for normal and tumor cells"24300711824306510.1016/j.dnarep.2007.12.0081601216710.1074/jbc.M503776200"Overexpression and hypomethylation of flap endonuclease 1 gene in breast and other cancers"29486711901081910.1158/1541-7786.MCR-08-02691687969310.1111/j.1464-410X.2006.06224.x157018302459040010.3892/ijmm.2014.16821592286310.1016/j.canlet.2004.10.035"Exploration of global gene expression patterns in pancreatic adenocarcinoma using cDNA microarrays"18512131265160710.1016/S0002-9440(10)63911-91456205410.1038/sj.onc.1206977"Hypomethylation of ETS transcription factor binding sites and upregulation of PARP1 expression in endometrial cancer"36663592376286710.1155/2013/946268"Poly (ADP-ribose) polymerase 1 transcriptional regulation: a novel crosstalk between histone modification H3K9ac and ETS1 motif hypomethylation in BRCA1-mutated ovarian cancer"39602092444842310.18632/oncotarget.1549"Promoter hypomethylation, especially around the E26 transformation-specific motif, and increased expression of poly (ADP-ribose) polymerase 1 in BRCA-mutated serous ovarian cancer"35993662344260510.1186/1471-2407-13-901177949310.1016/S1097-2765(01)00419-11646402210.1021/cr040481v""Great Oxidation Event: More oxygen through multicellularity.""1643755110.1002/ar.b.20089"The Evolutionary Dynamics of Digital and Nucleotide Codes: A Mutation Protection Perspective"3D structures of some DNA repair enzymesHuman DNA repair diseasesDNA RepairDNA Damage and DNA RepairSegmental ProgeriaDNA-damage repair; the good, the bad, and the uglyIDdna-repairD004260
Reparación do ADN
célulaADNxenomametabólicasluz ultravioletaradiaciónstranscriciónxenemutaciónsmitoseencimasreparación por escisión de basesescisión de nucleótidosreparación de discordanciasunión de extremos non homólogosunión de extremos mediada por microhomoloxíarecombinación homólogasíntese translesiónreplicación do ADNADN polimerasesadutos do ADNforcada de replicaciónxenesciclo celularmitoseapoptosehipótese de KnudsonenvellecementoproxeriasepixenéticosatmosferaoxíxenoPrecámbricobacteriasarqueaseucariotasbacteriófagosPremio Nobel de QuímicaTomas LindahlPaul ModrichAziz Sancarxenoma humanoxenes supresores de tumorestumorheteroxeneidade dos tumoresdobre héliceadutosdobre hélicesuperenroladohistonasdivisión celularfenotipoconversión xénicatautoméricoseucariotasADNnúcleomitocondriascromatinaciclo celularcromosomasdivisión celularhistonasADN mitocondrialnucleoideespecies reactivas do oxíxenoradicais libresATPfosforilación oxidativaencimasuperóxido dismutasecitoplasmasenescenciatelómerosADN non codificantelímite de Hayflickquiescenciaciclo celularsinalización celularcancromutacións8-hidroxidesoxiguanosinaadutoshidrocarburos aromáticos policíclicoscomplementariacromosoma homólogoxenotípicosneutrastecidoxenomacromátideenlaces fosfodiésterdímeros de pirimidinaluz UVpirimidínicasfotorreactivaciónfotoliaseluz azul/UVlonxitude de ondabacteriasfungosanimaisreparación por escisión de nucleótidosguaninaMGMTogtestequiométricacatalíticaresposta adaptativaalquilaciónmetilacióncitosinaadeninacomplementariounión de extremos non homólogosunión de extremos mediada por microhomoloxíarecombinación homólogaADN ligase IVADN ligasecofactorXRCC4deleciónstranslocaciónsreplicase o seu ADNeucariotasrecombinación V(D)Jreceptores de células BTsistema inmunitariovertebradosnucleaseMRE11PARP1FEN1XRCC1LIG3recombinación homólogaunión de extremos non homólogosdeleciónrecombinación homólogasobrecruzamento cromosómicomeiosecromátide irmáfase G2interfasereplicación do ADNcromosoma homólogoforcada de replicacióntopoisomerasessuperenrolamento do ADNADN polimerase Isobrecruzamentorecombinación homólogaRecAreplicación do ADNdímeros de timinasitios APADN polimerasessitios activospolimerasesmodificación postraducionalprocesividadePCNAPol ηPol ιfotodímero T^Tapareamentos de bases de Watson e Crickapareamento de bases de Hoogsteenmutación puntualpolimerasesplásmidoknockoutsSOSantíxeno nuclear de célula proliferanteubiquitinadoreplicación do ADNPol ζforcada de replicaciónradiación ionizanteraios ultravioletasproteínascarbohidratoslípidosARNadutos do ADNforcada de replicaciónapoptoseciclo celularmitosepuntos de controlciclo celularfase G1Sfase G2MquinasesATMATRfosforilantransdución de sinaisBRCA1MDC153BP1proteína quinasesfosfatidilinositol 3-quinaseantíxeno nuclear de célula proliferanteserina/treonina(S/T) quinasesataxia telanxiectasia mutadaevolucióncromosomasATMATRp53apoptoseinhibidor da quinase dependente de ciclinap21ciclinaquinase dependente de ciclinaresposta SOSexpresión xénicaLexARecAhomodímerorepresortranscricionaloperadorasdominioBacteriafilos bacterianosespiroquetasADN helicasehidrólise do ATPproteasedímerocaixas SOSpromotorespalindrómicaconservación de secuenciasfilos bacterianosresposta SOSeucariotaspunto de control do ciclo celularlévedosresposta ao shockreparación posreplicaciónreparación por escisión de nucleótidosARNmtelómeroslinfomasinfecciónscarcinóxenosradioactividaderestrición calóricataxa metabólicadose xénicanematodohomólogoreparación por escisión de basesADN mitocondrialpleiotropíaorixe biolóxica do envellecementoneuronasproxeriasenfermidades de envellecemento aceleradoanemia de Fanconicancro de mamacancro de coloncancro colorrectal non poliposo hereditarioNHEJrecombinación homólogaquimioterapiaradioterapiahematopoéticooncoxenesepixenéticasxenomasecuencia de nucleótidosmetilación do ADNmodificación de histonascromatinaHMGA2HMGA1microARNsdivisión celularinestabilidade xenéticareparación de discordancias no ADNrecombinacional homólogaaneuploidíastrastorno de deficiencia da reparación do ADNmutacióncodónMGMTmetilaciónpromotoracancros colorrectaisPMS2MLH1microARNmiR-155encimassíntese translesióncarcinoxéneserecombinacional homólogapresións selectivasunión de extremos mediada por microhomoloxíadeleciónFEN1endonucleasePARP1ETSendometrioMMEJcarcinoxéneseprocariotaseucariotasvirusbacteriófagosmotivos estruturaisevoluciónrexistro fósilPrecámbricoácidos nucleicosantepasado comúnatmosferaoxíxenofotosintéticoscatástrofe do oxíxenoradicais libresfosforilación oxidativaestrés oxidativomutaciónsliña xerminalgametosevoluciónpimpínsalelosevolucionabilidade
Reparación do ADN
Saltar ata a navegación
Saltar á procura
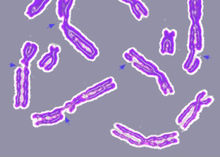
Danos no ADN que orixinaron múltiples roturas nos cromosomas (indicadas con frechas).
A reparación do ADN (ou reparación do DNA) é o conxunto de procesos polos cales unha célula detecta e corrixe os danos que se produciron nas moléculas de ADN nas que está codificado o seu xenoma. Nas células humanas, tanto as actividades metabólicas normais coma a influencia de factores ambientais como a luz ultravioleta (UV) e outras radiacións poden causar danos no ADN, que teñen como resultado a produción de lesións, que se estima que pode chegar ata aproximadamente un millón de pequenas lesións moleculares por célula e día.[1] Moitas destas lesións causan danos estruturais á molécula de ADN e poden alterar ou eliminar a capacidade da célula de realizar a transcrición dun xene que foi afectado pola lesión. Outras lesións inducen mutacións potencialmente nocivas no xenoma celular, que poden afectar a supervivencia das súas células fillas despois da mitose ou orixinar doenzas. Como consecuencia, o proceso de reparación do ADN está activo constantemente para responder a estes danos na estrutura do ADN.
A célula ten diversos mecanismos para reparar os danos no ADN, que poden necesitar un molde para guiarse na reparación e resintetizar as partes danadas ou non necesitalo, e poden realizar a reparación con gran precisión ou de forma máis imprecisa (o ADN non queda igual ao orixinal), e mesmo poden perderse segmentos de ADN. Estes mecanismos son: (1) reversión directa, que utiliza encimas que non precisan dun molde; (2) reparación por escisión, que utiliza a outra febra do ADN como molde para reparar roturas de febra simple, e pode realizarse por reparación por escisión de bases, por escisión de nucleótidos ou por reparación de discordancias; (3) reparación de roturas de dobre febra, que pode realizarse por unión de extremos non homólogos, unión de extremos mediada por microhomoloxía ou por recombinación homóloga, e (4) síntese translesión, que funciona durante a replicación do ADN utilizando unhas ADN polimerases especiais.
A acumulación de danos, especificamente de roturas de dobre febra ou adutos do ADN que fan que quede atascada a forcada de replicación, están entre os sinais estimuladores que se sabe desencadean unha resposta global aos danos no ADN, dirixida á preservación das propias células, que activa moitas vías de reparación macromolecular, sorteo dos puntos lesionados, tolerancia, ou apoptose, cuxas características comúns son a indución de moitos xenes, detención do ciclo celular e inhibición da mitose.
Cando os procesos normais de reparación fallan, e cando non ocorre a apoptose celular (morte das células danadas), pode ocorrer un dano no ADN irreparable, como por exemplo roturas de dobre febra e enlaces cruzados no ADN, é dicir, enlaces cruzados entre as dúas febras (ou ICLs, polas súas siglas en inglés de Interstrand CrossLinks).[2][3] Isto pode finalmente orixinar tumores malignos ou cancros segundo a hipótese de Knudson.
A taxa de reparación do ADN depende de moitos factores, como o tipo celular, a idade da célula e o ambiente extracelular. Unha célula que acumulara unha gran cantidade de danos no ADN, ou que xa non poida reparar de forma eficaz ditos danos, pode entrar en tres posibles estadios, que son:[4]
- Un estadio irreversible de dormencia, chamado senescencia.
- O suicidio celular, tamén chamado apoptose ou morte celular programada.
- Divisións celulares non reguladas, que poden levar a formación dun tumor que sexa canceroso.
A capacidade de reparación do ADN dunha célula é vital para conservar a integridade do seu xenoma e, por tanto, para o normal funcionamento de todo o organismo. Moitos xenes que se viu inicialmente que influían na duración da vida, comprobouse despois que estaban implicados na reparación e protección do ADN.[5] Os danos no ADN propóñense como unha das causas principais do envellecemento. Diversos trastornos xenéticos hereditarios, como as proxerias, débense a alteracións dos xenes que interveñen na reparación do ADN.
Os estudos epixenéticos mostraron que as alteracións epixenéticas nos xenes de reparación do ADN teñen unha influencia esencial na carcinoxénese.
Os mecanismos de reparación do ADN tiveron que xurdir cando na Terra se orixinou unha atmosfera rica en oxíxeno no Precámbrico, que causaba danos oxidativos ao ADN. Os procesos básicos desta reparación están moi conservados en todos os dominios da vida: bacterias, arqueas, eucariotas, e mesmo tamén en virus bacteriófagos.
 Play media
Play mediaPaul Modrich
Outorgouse o Premio Nobel de Química de 2015 a Tomas Lindahl, Paul Modrich e Aziz Sancar polos seus traballos sobre os mecanismos moleculares dos procesos de reparación do ADN.[6][7]
Índice
1 Danos no ADN
1.1 Fontes dos danos
1.2 Tipos de danos
1.3 Danos no ADN nuclear e mitocondrial
1.4 Senescencia e apoptose
1.5 Danos no ADN e mutacións
2 Mecanismos de reparación do ADN
2.1 Reversión directa
2.2 Danos de febra simple
2.3 Roturas de dobre febra
2.4 Síntese translesión
3 Resposta global aos danos no ADN
3.1 Puntos de control de danos no ADN
3.2 A resposta SOS procariota
3.3 Respostas transcricionais eucariotas a danos no ADN
4 Reparación do ADN e envellecemento
4.1 Efectos patolóxicos dunha deficiente reparación do ADN
4.2 Lonxevidade e restrición calórica
5 Medicina e modulación da reparación do ADN
5.1 Trastornos de reparación do ADN hereditarios
6 Reparación do ADN e cancro
6.1 Defectos de reparación do ADN epixenéticos no cancro
6.2 Frecuencias de epimutacións en xenes para a reparación do ADN
7 Reparación do ADN e evolución
7.1 Taxa de cambio evolutivo
8 Notas
9 Véxase tamén
9.1 Outros artigos
9.2 Ligazóns externas
Danos no ADN |
Os danos no ADN, tanto os debidos a factores ambientais coma a procesos metabólicos celulares, ocorren, segundo as estimacións, cunha frecuencia de entre 10 000 e 1 000 000 de lesións moleculares por célula e día.[1][8] Aínda que estes danos afectan só o 0,000165% dos 3 000 millóns de pares de bases do xenoma humano, as lesións non reparadas en xenes críticos (como os xenes supresores de tumores[9]) poden impedir que a célula leve a cabo a súa función e incrementan apreciablemente a probabilidade de que se orixine un tumor e contribúe á heteroxeneidade dos tumores.[10]
A maioría dos danos no ADN afectan a estrutura primaria da dobre hélice; é dicir, as propias bases son modificadas quimicamente. Estas modificacións poden á súa vez alterar a estrutura helicoidal das moléculas[11] ao introducir enlaces químicos non nativos ou adutos voluminosos que non encaixan ben na estrutura da dobre hélice estándar.[12] O ADN está superenrolado e asociado a proteínas de "empaquetamento" chamadas histonas (en eucariotas), e ambas as estruturas son vulnerables aos efectos dos danos no ADN, e deben ser reparadas.[13] A propia modificación de histonas forma parte dos mecanismos de recoñecemento dos danos e reparación do ADN.[14]
Fontes dos danos |
Os danos no ADN poden ser subdivididos en dous tipos principais:[15]
- Danos endóxenos orixinados por causas internas como:
- Mutacións espontáneas como as causadas polo ataque de especies reactivas do oxíxeno producidas a partir de subprodutos metabólicos normais, especialmente no proceso de desaminación oxidativa.[16]
- Tamén inclúe os erros de replicación.
- Danos exóxenos causados por axentes externos como:
- Radiación ultravioleta [UV 200-400 nm] do sol.[16]
- Radiacións doutras frecuencias, como raios X e gamma.[16]
Hidrólise[17] ou alteración térmica.[18]- Certas toxinas de plantas.[19]
Compostos químicos mutaxénicos artificiais, especialmente os aromáticos, que actúan como axentes intercalantes do ADN.[20]
Virus.[21]
A replicación do ADN danado antes da división celular pode causar a incorporación de bases incorrectas na cadea complementaria do ADN en fronte das bases danadas. As células fillas que herdan estas bases incorrectas levan mutacións, polo que a secuencia de ADN orixinal é irrecuperable, agás no raro caso de que se produza unha mutación retrógrada (é dicir, unha mutación puntual que restaura a secuencia orixinal e, por tanto, o fenotipo orixinal [22]), por exemplo por conversión xénica.
Tipos de danos |
Hai varios tipos de danos no ADN debidos a procesos celulares endóxenos:
Oxidación de bases (por exemplo, xeración de 8-oxo-7,8-dihidroguanina) e xeración de interrupcións da febra de ADN causadas por especies reactivas do oxíxeno.[23][24]
Alquilación de bases (usualmente metilación), como a formación de 7-metilguanina, 1-metiladenina, 6-O-metilguanina.[25]
Hidrólise de bases, como a desaminación, despurinación e despirimidinación.[26]- Formación de adutos voluminosos (como os adutos benzo[a]pireno diol epóxido-dG ou aristolactam I-dA).[15][27]
- Discordancia na complementariedade de bases, debido a erros na replicación do ADN, na cal se coloca nunha febra de ADN de nova formación unha base errada, ou se omite unha base ou é inserida indebidamente unha base extra.[15]
- Danos de monoadutos causados por cambios nunha soa base nitroxenada do ADN.[28]
- Danos de diadutos.[28]
Os danos causados por axentes exóxenos prodúcense de moitas formas. Algúns exemplos son:
- A luz UV-B causa a formación de enlaces cruzados entre bases timina e citosina adxacentes creando dímeros de pirimidina. Isto denomínase danos directos ao ADN.[29]
- A luz UV-A crea principalmente radicais libres. Os danos causados polos radicais libres denomínase danos indirectos ao ADN.[30]
- As radiacións ionizantes como as xeradas pola radioactividade ou os raios cósmicos causan roturas nas febras do ADN. A radiación ionizante de nivel intermedio pode inducir danos irreparables no ADN (que causan erros na replicación e transcrición que son un paso necesario para a formación de neoplasias ou poden desencadear interaccións virais) que orixinan un envellecemento prematuro e cancros.[31]
- A alteración térmica a temperaturas elevadas incrementa o grao de despurinación (perda de bases púricas no ADN) e roturas dunha febra. Por exemplo, a despurinación hidrolítica dáse nas bacterias termófilas, que crecen en fontes termais a 40-80 °C.[18][32] A taxa de despurinación (300 residuos de purina por xenoma e por xeración) é demasiado alta nestas especies como para ser reparada pola maquinaria normal de reparación, polo que non se pode descartar a posibilidade de que sexa unha resposta adaptativa.
- Compostos químicos industriais como o cloruro de vinilo[33] e o peróxido de hidróxeno[34], e compostos químicos ambientais como hidrocarburos aromáticos policíclicos[35] que se encontran no fume, feluxe e alcatrán crean unha gran diversidade de adutos do ADN, como etenobases, bases oxidadas, fosfotriésteres alquilados e enlaces cruzados no ADN, entre outros.[36]
Outra maneira de clasificar os danos é en inducidos e espontáneos. Os danos por luz UV, alquilación/metilación, por raios X e danos oxidativos son exemplos de danos inducidos. Os danos espontáneos inclúen a perda dunha base, desaminación, deformacións nos aneis de azucres e cambios tautoméricos.[15]
Danos no ADN nuclear e mitocondrial |
Nas células humanas, e, en xeral, nas células eucariotas, o ADN encóntrase en dúas localizacións celulares: dentro do núcleo e dentro das mitocondrias. O ADN nuclear (ADNn) está en forma de cromatina durante os estados non replicativos do ciclo celular e condénsase en estruturas agregadas chamadas cromosomas durante a división celular. En ambos os estados o ADN está moi compactado e enrolado arredor de grupos de proteínas histonas. Cando unha célula necesita expresar a información xenética codificada no seu ADNn a rexión cromatínica implicada é desenredada, e os xenes localizados alí son expresados, e despois a rexión volve condensarse á súa conformación anterior.[37] O ADN mitocondrial (ADNmt) está localizado dentro das mitocondrias, é circular e bicatenario, del hai moitas copias, e tamén está estreitamente asociado con varias proteínas coas que forma un complexo chamado nucleoide. Dentro da mitocondria as especies reactivas do oxíxeno (ROS), ou radicais libres, que son subprodutos da constante formación de ATP por fosforilación oxidativa, crean unha contorna moi oxidante que se sabe produce danos no ADNmt. Un encima fundamental para contrarrestar a toxicidade destas especies é a superóxido dismutase, a cal está presente tanto en mitocondrias coma no citoplasma das células eucariotas.[38][39][40]
Senescencia e apoptose |
A senescencia, que é un proceso irreversible no cal a célula xa non se divide, é unha resposta protectora ao acurtamento dos telómeros (extremos cromosómicos). Os telómeros son longas rexións formadas por ADN non codificante repetitivo que forman unha especie de tope no extremo dos cromosomas, pero que sofren unha degradación parcial (polo que se acurtan) cada vez que a célula se divide (ver límite de Hayflick).[41] O estado de quiescencia é un estado irreversible de dormencia celular que non está relacionado con danos no ADN (ver ciclo celular). A senescencia nas células pode servir como unha alternativa funcional á apoptose en casos onde cómpre a presenza física dunha célula por razóns espaciais para o organismo,[42] o cal serve como mecanismo de "último recurso" para impedir que unha célula con ADN danado se replique inapropriadamente en ausencia dunha sinalización celular que indique crecemento. As división celulares non reguladas poden levar á formación dun tumor (ver cancro), potencialmente mortal para o organismo. Xa que logo, a indución da senescencia e a apoptose considérase que é parte dunha estratexia de protección contra o cancro.[43]
Danos no ADN e mutacións |
Cómpre distinguir entre danos no ADN e mutacións, que son os dous maiores tipos de erros que se poden atopar no ADN, pero que son moi diferentes. Os danos poden causar mutacións pero non son exactamente o mesmo. Os danos son anormalidades físicas no ADN, como roturas de dobre febra ou dunha febra, presenza de residuos alterados como os de 8-hidroxidesoxiguanosina (ou 8-oxo-2'-desoxiguanosina) e adutos de hidrocarburos aromáticos policíclicos. Os danos no ADN poden ser recoñecidos por encimas, que os poden reparar se hai información redundante, como unha secuencia non danada na febra complementaria do ADN ou no cromosoma homólogo, que pode ser copiada. Se unha célula conserva o ADN danado, isto pode impedir a transcrición dalgún xene, que nunca se traducirá a proteínas. A replicación pode tamén ser bloqueada ou a célula pode morrer.[15][44]
A diferenza dos danos no ADN, unha mutación é un cambio na secuencia de bases do ADN ou perda de segmentos de ADN, que orixina cambios xenotípicos. Unha mutación non pode ser recoñecida por encimas unha vez que o cambio de bases está presente en ambas as febras do ADN, polo que unha mutación non se pode reparar. A nivel celular, as mutacións poden causar alteracións no funcionamento das proteínas e na regulación. As mutacións son tamén replicadas cando a célula se divide. Nunha poboación de células, as células mutantes incrementan ou diminúen a súa frecuencia segundo os efectos que a mutación teña sobre a súa capacidade de sobrevivir e reproducirse. Aínda que sexan cousas distintas, os danos no ADN e as mutacións están relacionados porque os danos no ADN a miúdo causan erros na síntese de ADN durante a replicación ou reparación; estes erros son unha fonte importante de mutacións.[45]
Dadas as propiedades que teñen os danos no ADN e as mutacións, os danos no ADN son un especial problema en células que non se dividen ou que que se dividen lentamente, nas que os danos non reparados tenderán a acumularse co tempo. Por outra parte, en células que se dividen rapidamente, os danos no ADN non reparados que non matan a célula ao bloquearen a replicación tenderán a causar erros de replicación e, por tanto, mutacións. A gran maioría das mutacións que non son neutras nos seus efectos, son deletéreas para a supervivencia da célula. Así, nunha poboación de células que compoñen un tecido con células en replicación, as células mutantes tenderán a perderse. Porén, certas mutacións infrecuentes que proporcionan unha vantaxe para a supervivencia tenderán a expandirse clonalmente a expensas das células veciñas normais do tecido. Esta vantaxe para a célula é desvantaxosa para o conxunto do organismo, porque esas células mutantes poden dar orixe a un cancro. Deste xeito, como os danos no ADN en células que se dividen frecuentemente dan lugar a mutacións, son unha causa importante de cancro. A diferenza do anterior, os danos no ADN en células que non se dividen frecuentemente son probablemente unha causa importante do envellecemento.[46]
Mecanismos de reparación do ADN |
As células non poden funcionar se os danos no ADN afectan a integridade e accesibilidade de información esencial do xenoma, pero as células aparentemente seguen sendo funcionais cando os chamados xenes "non esenciais" se perden ou quedan danados. Dependendo do tipo de dano inflixido á estrutura de dobre hélice do ADN, a célula pode aplicar varias estratexias de reparación que evolucionaron para restaurar a información perdida. Se é posible, as células usan a febra complementaria non modificada do ADN ou a cromátide irmá como molde para recuperar a información orixinal. Cando non teñen acceso a un molde, as células usan como último recurso un mecanismo de recuperación tendente ao erro chamado síntese translesión.[47][48]
Os danos no ADN alteran a configuración espacial da hélice, e ditas alteracións pode detectalas a célula. Unha vez que se localiza o dano, únense moléculas de reparación do ADN específicas ao sitio (ou preto do sitio) onde se produciu o dano, o que induce a outras moléculas a unirse tamén e formar un complexo que permite que a reparación teña lugar.[48]
Reversión directa |
As células poden eliminar tres tipos de danos no seu ADN reverténdoos quimicamente. Estes mecanismos non requiren a utilización dunha secuencia molde, xa que os tipos de danos que se contrarrestan poden ocorrer só nunha das catro bases. Estes mecanismos de reversión directa son específicos do tipo de danos producidos e non implican a rotura do esqueleto de enlaces fosfodiéster. A formación de dímeros de pirimidina por irradiación con luz UV orixina enlaces covalentes anormais entre bases pirimidínicas adxacentes. O proceso de fotorreactivación reverte directamente este dano pola acción do encima fotoliase, cuxa activación é obrigadamente dependente da enerxía absorbida da luz azul/UV (lonxitude de onda de 300-500 nm) para promover a catálise.[49] A fotoliase é un vello encima presente en bacterias, fungos e animais, pero xa non funciona nos humanos,[50] que teñen no seu lugar o mecanismo de reparación por escisión de nucleótidos para reparar os danos causados pola irradiación UV. Outro tipo de danos, a metilación das bases guanina, revértese directamente pola intervención da proteína metil guanina metil transferase (MGMT), que ten un equivalente bacteriano chamado ogt. Este é un proceso caro para a célula porque cada molécula de MGMT pode utilizarse unha soa vez, é dicir, a reacción é estequiométrica en lugar de catalítica.[51] Unha resposta xeneralizada aos axentes metilantes en bacterias é a denominada resposta adaptativa, que dá certo nivel de resistencia aos axentes alquilantes despois dunha exposición continuada por regulación á alza dos encimas de reparación da alquilación.[52] O terceiro tipo de dano no ADN que poden reverter as células é certa metilación das bases citosina e adenina.
Danos de febra simple |
Cando só unha das dúas febras da dobre hélice ten defectos, a outra febra pode utilizarse como molde para guiar a corrección da febra danada. Para reparar os danos dunha das dúas febras apareadas do ADN, hai varios mecanismos de reparación por escisión que retiran o nucleótido danado e substitúeno por outro nucleótido correcto que sexa complementario co que se encontra na febra do ADN non alterada.[51] Estes mecanismos son:

Estrutura do encima de reparación por escisión de bases uracilo-ADN glicosilase. O residuo de uracilo móstrase en amarelo.
- A reparación por escisión de bases (BER) repara os danos que afectan a unha soa base nitroxenada mediante a actuación de encimas chamados glicosilases.[53] Estes encimas eliminan unha soa base nitroxenada, creando un sitio purínico ou apurínico (sitio AP).[53] Os encimas AP endonucleases fan unha amosega ou corte no esqueleto da molécula de ADN danada no sitio AP. Despois, a ADN polimerase elimina a rexión danada utilizando a súa actividade exonuclease 5'-3' e sintetiza correctamente a nova febra utilizando a febra complementaria como molde.[53]
- A reparación por escisión de nucleótidos (NER) repara o ADN danado, que xeralmente consiste nun dano voluminoso que distorsiona a hélice, como pode ser unha dimerización de pirimidina causada pola luz UV. As rexións danadas elimínanse en febras de 12 a 24 nucleótidos de longo nun proceso en tres fases, que consiste no recoñecemento dos danos, a escisión do ADN danado tanto augas arriba coma augas abaixo do punto do dano por medio dunha endonuclease, e a resíntese da rexión de ADN eliminada.[54] A NER é un mecanismo de reparación moi conservado evolutivamente e utilízase en case todas as células eucariotas e procariotas.[54] En procariotas, a NER realízana encimas Uvr.[54] En eucariotas, están implicadas moitas máis proteínas, aínda que a estratexia xeral é a mesma.[54]
- Os sistemas de reparación de discordancias (MMR) na complementariedade de bases están presentes esencialmente en todas as células para corrixir erros que non son corrixidos por corrección de probas. Estes sistemas constan de polo menos dúas proteínas. Unha detecta a discordancia na complementariedade, e a outra recruta unha endonuclease que escinde a febra de ADN de nova síntese preto da rexión onde se produciu o dano. En E. coli, as proteínas implicadas son as proteínas de clase Mut. Isto vai seguido da eliminación da rexión danada por unha exonuclease, a resíntese pola ADN polimerase e a selaxe do corte por unha ADN ligase.[55]
Roturas de dobre febra |
As roturas de dobre febra, nas cales nun punto hai un corte que afecta a ambas as febras da dobre hélice, son especialmente perigosas para a célula porque poden orixinar rearranxos xenómicos. Hai tres mecanismos que reparan as roturas de dobre febra (en inglés DSBs): unión de extremos non homólogos (en inglés NHEJ), unión de extremos mediada por microhomoloxía (en inglés MMEJ) e recombinación homóloga.[51] O investigador P. V. Acharya sinalou que as roturas de dobre febra e unha "unión por enlaces cruzados que ligue ambas as febras no mesmo punto é irreparable porque ningunha das febras pode servir como molde para facer a reparación. A célula morrerá na seguinte mitose ou en raros casos, mutará."[2][3]

A ADN ligase, que se mostra na parte superior reparando un dano cromosómico, é un encima que une nucleótidos rotos ao catalizar a formación dun enlace éster internucleotídico entre o esqueleto de fosfatos da molécula e os desoxirribonucleótidos.
Na unión de extremos non homólogos (NHEJ), a ADN ligase IV, unha ADN ligase especializada que forma un complexo co cofactor XRCC4, encárgase de unir directamente os dous extremos.[56] Para guiar unha reparación exacta, a NHEJ depende de curtas secuencias homólogas chamadas microhomoloxías presentes en colas de febra simple dos extremos do ADN que van ser unidos. Se estas colas que sobresaen son compatibles en bases, a reparación é xeralmente exacta.[57][58][59][60] A NHEJ pode tamén introducir mutacións durante a reparación. A perda de nucleótidos danados no sitio de rotura pode orixinar delecións, e a unión de extremos discordantes (non complementarios) causa translocacións. A NHEJ é especialmente importante antes de que a célula replicase o seu ADN, xa que non hai dispoñible un molde para a reparación por recombinación homóloga. Hai vías da NHEJ alternativas ou redundantes (de "back up") en eucariotas superiores.[61] Ademais do seu papel como vixilante do xenoma, a NHEJ é necesaria para unir roturas de dobre febra que acaban en forquita inducidas durante a recombinación V(D)J, que é o proceso que xera diversidade nos receptores de células B e T no sistema inmunitario de vertebrados.[62]
A MMEJ comeza cunha resección de extremos de curto rango feita pola nuclease MRE11 a cada lado dunha rotura de dobre febra para revelar rexións de microhomoloxía.[63] Nos seguintes pasos[64] é necesario a PARP1 e pode ser un paso inicial na MMEJ. Hai un emparellamento de rexións de microhomoloxía seguida do recrutamento da endonuclease FEN1 (flap structure-specific endonuclease 1) para que elimine as solapas que sobresaen. Despois prodúcese o recrutamento de XRCC1–LIG3 no sitio para ligar os extremos do ADN, o que orixina un ADN intacto.
As roturas de dobre hélice no ADN en células de mamíferos son reparadas primariamente por recombinación homóloga (HR) e unión de extremos non homólogos (NHEJ).[65] Nun sistema in vitro, a MMEJ ten lugar nas células de mamíferos a un nivel equivalente a do 10 ao 20% da HR cando están dispoñibles ambos os mecanismos.[63] A MMEJ está sempre acompañada dunha deleción, polo que a MMEJ é unha vía mutaxénica de reparación do ADN.[66]
A recombinación homóloga require a presenza dunha secuencia idéntica ou case idéntica para usala como molde para a reparación da rotura. A maquinaria encimática responsable deste proceso de reparación é case idéntico á responsable do sobrecruzamento cromosómico que ten lugar durante a meiose. Esta vía permite que os cromosomas danados sexan reparados usando como molde unha cromátide irmá (dispoñible na fase G2 da interfase despois da replicación do ADN) ou un cromosoma homólogo. As roturas de dobre febra causadas pola maquinaria de replicación que intenta sintetizar a través dunha rotura de febra simple ou lesión non reparada causan o colapso da forcada de replicación e son reparadas normalmente por recombinación.[67][68]
As topoisomerases introducen roturas tanto de febra simple coma dobre cando orixinan o cambio do estado de superenrolamento do ADN, o cal é especialmente común en rexións preto da forcada de replicación. Esas roturas non se consideran danos no ADN porque son un intermediario natural no mecanismo bioquímico das topoisomerases e son inmediatamente reparadas polos encimas que as crearon.[69]
Un equipo de investigadores croatas e franceses expuxeron a radiacións a bacteria Deinococcus radiodurans orixinando moitos fragmentos do seu xenoma para estudaren o mecanismo de reparación das roturas de dobre febra do ADN nese organismo. Atoparon un novo mecanismo de reparación, chamado "annealing de febras dependente de síntese estendida" (ou ESDA, do inglés extended synthesis-dependent strand annealing), na cal cómpren polo menos dúas copias do xenoma, con roturas aleatorias no ADN. Os fragmentos con homoloxías que se solapan utilízanse para sintetizar as febras complementarias que faltan, o que é catalizado pola ADN polimerase I, que estende a febra máis do normal. Os fragmentos así formados teñen a particularidade de que presentan extremos cohesivos, que facilmente poden facer o annealing (unirse por apareamento complementario de bases) e unirse a outros fragmentos complementarios de ADN do conxunto, orixinando fragmentos intermediarios máis grandes. No paso final para formar cromosomas circulares hai un sobrecruzamento por medio de recombinación homóloga dependente de RecA.[70]
Síntese translesión |
A síntese translesión (TLS) é un proceso que permite tolerar danos no ADN para que a maquinaria de replicación do ADN poida replicar máis alá dos puntos onde hai lesión no ADN, como dímeros de timina ou sitios AP.[71] Funciona trocando as ADN polimerases normais por polimerases translesión especializadas (é dicir, as ADN polimerases IV ou V, pertencentes á familia de polimerases Y), que adoitan ter sitios activos máis grandes que poden facilitar a inserción de bases situadas en fronte de nucleótidos danados. Este cambio de polimerases crese que é mediado, entre outros factores, pola modificación postraducional do factor de procesividade da replicación PCNA. As polimerases de síntese translesión adoitan ter unha baixa fidelidade (alta propensión a inserir bases erradas) ao copiaren moldes non danados en relación coas polimerases normais. Porén, moitas son extremadamente eficaces á hora de inserir as bases correctas en fronte dos lugares onde hai tipos específicos de danos. Por exemplo, a Pol η (eta) realiza un rodeo ou bypass libre de erros de lesións inducidas por irradiación ultravioleta, mentres que a Pol ι (iota) introduce mutacións neses sitios. A Pol η engade a primeira adenina por medio do fotodímero T^T usando apareamentos de bases de Watson e Crick e a segunda adenina é engadida na súa conformación syn usando apareamento de bases de Hoogsteen.[72][73]
Desde unha perspectiva celular, o risco de introducir unha mutación puntual durante a síntese translesión pode ser preferible a utilizar mecanismos máis drásticos de reparación do ADN, que poden causar grandes aberracións cromosómicas ou a morte celular. En resumo, o proceso implica polimerases especializadas que reparan ou dan un rodeo (bypass) pola lesión en lugares onde a replicación do ADN queda atascada. Por exemplo, a ADN polimerase eta humana pode superar os puntos con lesións do ADN complexas como poden ser enlaces cruzados intrafebra guanina-timina (G[8,5-Me]T).[74] P. Raychaudhury e A. Basu[75] estudaron a toxicidade e a mutaxénese da mesma lesión en E. coli replicando un plásmido coa modificación G[8,5-Me]T en E. coli con knockouts específicos de ADN. A viabilidade era moi baixa nunha cepa que carecía de pol II, pol IV e pol V, que son as tres ADN polimerases inducibles SOS, o que indica que a síntese translesión a realizan principalmente estas ADN polimerases especializadas.
O antíxeno nuclear de célula proliferante (PCNA) proporciona unha plataforma de bypass para estas polimerases. En circunstancias normais, a replicación do ADN faise co PCNA unido a polimerases. Nun sitio de lesión, o PCNA é ubiquitinado, ou modificado, polas proteínas RAD6/RAD18 para proporcionar unha plataforma para que as polimerases especializadas sorteen o punto da lesión e recomecen a replicación do ADN.[76][77] Despois da síntese translesión, cómpre facer unha extensión da febra. Esta extensión pode levala a cabo unha polimerase replicativa se a síntese translesión non presenta erros, como no caso da Pol η, aínda que se a síntese translesión orixina un erro no apareamento de bases, necesítase unha polimerase especializada para facer esta extensión, que é a Pol ζ. A Pol ζ ten a característica exclusiva de que pode estender discordancias no apareamento de bases terminais, mentres que outras polimerases máis procesivas non poden. Así que cando se encontra unha lesión e a forcada de replicación queda detida, o PCNA cambia de estar unido a unha polimerase procesiva a estalo a unha polimerase de síntese translesión (TLS) como Pol ι para reparar a lesión, despois o PCNA pode cambiar á Pol ζ para estender a discordancia na complementariedade de bases, e finalmente, o PCNA cambia de novo á polimerase procesiva para continuar coa replicación.[73]
Resposta global aos danos no ADN |
As células expostas á radiación ionizante, raios ultravioletas ou certos compostos químicos sofren en moitos sitios do seu ADN lesións voluminosas e roturas de dobre febra. Ademais, os axentes que danan o ADN poden danar tamén outras moléculas como proteínas, carbohidratos, lípidos e ARN. A acumulación de danos, especificamente de roturas de dobre febra ou adutos do ADN que fan que quede atascada a forcada de replicación, están entre os sinais estimuladores que se sabe desencadean unha resposta global aos danos no ADN.[78] A resposta global aos danos é unha acción dirixida á preservación das propias células e activa moitas vías de reparación macromolecular, sorteo dos puntos lesionados, tolerancia, ou apoptose. As características comúns da resposta global son a indución de moitos xenes, detención do ciclo celular e inhibición da mitose.
Puntos de control de danos no ADN |
Cando se producen danos no ADN, actívanse os puntos de control (checkpoints) do ciclo celular.[79] A activación dos puntos de control detén o ciclo celular e dálle tempo á célula a reparar os danos antes de que esta continúe a dividirse. Os puntos de control de danos no ADN funcionan nos límites entre fase G1/S e fase G2/M. Tamén existe un punto de control dentro da fase S. A activación de puntos de control está controlada por dúas quinases mestras, a ATM e ATR. A quinase ATM responde ás roturas de dobre febra do ADN e altera a estrutura da cromatina,[80] mentres que a ATR responde principalmente á presenza de forcadas de replicación detidas. Estas quinases fosforilan dianas situadas augas abaixo nunha fervenza de transdución de sinais, o que finalmente fai que o ciclo celular se deteña. Tamén se identificou unha clase de proteínas mediadoras do punto de control, como BRCA1, MDC1 e 53BP1.[81] Estas proteínas parecen ser necesarias para transmitir o sinal de activación do punto de control a proteínas situadas augas abaixo da vía.
As proteínas dos puntos de control poden dividirse en catro grupos: proteína quinases do tipo PI3K (fosfatidilinositol 3-quinase), grupo similar a PCNA (antíxeno nuclear de célula proliferante), e dúas serina/treonina(S/T) quinases e os seus adaptadores. Son esenciais en todas as respostas de puntos de control inducidos por danos no ADN un par de grandes proteína quinases que pertencen ao primeiro grupo das proteína quinases de tipo PI3K, chamadas quinases ATM (ataxia telanxiectasia mutada) e ATR (relacionadas coa ataxia e rad), cuxas secuencias e funcións foron ben conservadas na evolución. Todas as respostas a danos no ADN requiren a ATM ou a ATR porque estas teñen a capacidade de unirse aos cromosomas no sitio onde se produciu o dano no ADN, xunto con proteínas accesorias que son plataformas sobre as cales poden ensamblarse os compoñentes da resposta aos danos do ADN e os complexos de reparación do ADN.[82]
Unha importante diana situada augas abaixo da ATM e a ATR é p53, xa que esta proteína cómpre para inducir a apoptose despois dun dano no ADN.[83] O inhibidor da quinase dependente de ciclina p21 é inducido tanto por mecanismos dependentes de p53 coma independentes de p53 e pode deter o ciclo celular nos puntos de control G1/S e G2/M desactivando os complexos ciclina/quinase dependente de ciclina.[84]
A resposta SOS procariota |
A resposta SOS consiste nos cambios na expresión xénica que se producen en Escherichia coli e outras bacterias en resposta a grandes danos no ADN. O sistema SOS procariota é regulado por dúas proteínas clave: LexA e RecA. O homodímero LexA é un represor transcricional que se une a secuencias operadoras normalmente denominadas caixas SOS. En Escherichia coli sábese que LexA regula a transcrición de aproximadamente 48 xenes incluíndo os xenes lexA e recA.[85] A resposta SOS está moi estendida entre o dominio Bacteria, mais case sempre está ausente en certos filos bacterianos, como o das espiroquetas.[86] Os sinais celulares máis comúns que activan as respostas SOS son rexións de ADN monocatenario, que se orixinan en forcadas de replicación detidas ou roturas de dobre febra, que son procesadas pola ADN helicase para separar as dúas febras do ADN.[78] No paso de iniciación, a proteína RecA únese ao ADN monocatenario nunha reacción impulsada pola hidrólise do ATP, creando filamentos RecA–ADN monocatenario. Os filamentos RecA–ADN monocatenario activan a actividade de autoprotease de LexA, o cal leva finalmente a que se produza a escisión do dímero LexA, seguida da degradación de LexA. A perda do represor LexA induce a transcrición dos xenes SOS e permite unha indución de sinais, inhibición da división celular e un incremento dos niveis de proteínas responsables do procesamento dos danos.
En Escherichia coli, as caixas SOS teñen secuencias de 20 nucleótidos de longo situadas preto de promotores con estrutura palindrómica e un alto grao de conservación de secuencias. Noutras clases e filos bacterianos, a secuencia das caixas SOS varía considerablemente, e presenta diferente lonxitude e composición, pero está sempre moi conservada e é un dos sinais curtos máis fortes do xenoma.[86] O alto contido informativo das caixas SOS permite a unión diferencial de LexA a diferentes promotores e permite unha correcta temporalización da resposta SOS. Os xenes de reparación de lesións son inducidos ao comezo da resposta SOS. As polimerases translesión con tendencia ao erro, por exemplo, UmuCD'2 (tamén chamada ADN polimerase V), son inducidas posteriormente como un último recurso.[87] Unha vez que se reparan ou sortean os danos no ADN utilizando polimerases ou por medio de recombinación, a cantidade de ADN monocatenario nas células baixa, ao diminuír a cantidade de filamentos RecA descende a actividade de escisión do homodímero LexA, que despois se une ás caixas SOS preto dos promotores e restaura a expresión xénica normal.
Respostas transcricionais eucariotas a danos no ADN |
As células eucariotas expostas a axentes que danan o ADN tamén activan vías defensivas importantes ao induciren moitas proteínas implicadas na reparación do ADN, o control do punto de control do ciclo celular e o tráfico e degradación de proteínas. Esta resposta transcricional en todo o xenoma é moi complexa e está moi regulada, o que permite unha resposta coordinada aos danos. A exposición de células de lévedos Saccharomyces cerevisiae a axentes que danan o ADN dá lugar a perfís transcricionais solapados pero diferentes. As semellanzas coa resposta ao shock ambiental indican que existe unha vía de resposta ao estrés global xeral a nivel de activación transcricional. En contraste, diferentes tipos de células humanas responden aos danos de xeito diferente, o que indica a ausencia dunha resposta global común. A probable explicación desta diferenza entre os lévedos e as células humanas pode estar na heteroxeneidade das células de mamífero. Nun animal hai diferentes tipos de células que están distribuídas en distintos órganos nos que evolucionaron diferentes sensibilidades aos danos no ADN.[88]
En xeral a resposta global aos danos no ADN implica a expresión de múltiples xenes responsables da reparación posreplicación, a recombinación homóloga, a reparación por escisión de nucleótidos, o punto de control de danos no ADN, a activación transcricional global, xenes que controlan a degradación do ARNm e moitos outros. Cando unha célula sofre unha gran cantidade de danos, esta opta entre sufrir apoptose e morrer ou sobrevivir a costa de vivir cun xenoma modificado. Un incremento na tolerancia aos danos pode levar a un incremento da proporción de supervivencia que permitirá a acumulación dunha maior cantidade de mutacións. As Rev1 de lévedos e a polimerase η humana son membros da familia Y de ADN polimerases translesión presentes durante a resposta global aos danos no ADN, e son responsables dunha potenciación da mutaxénese durante a resposta global aos danos no ADN en eucariotas.[78]
Reparación do ADN e envellecemento |
Efectos patolóxicos dunha deficiente reparación do ADN |

A taxa de reparación do ADN é un importante determinante da patoloxía celular.

A maioría dos xenes que inflúen na duración da vida afectan á taxa de danos no ADN.
Os animais experimentais que teñen deficiencias xenéticas na súa reparación do ADN adoitan ter unha duración da vida menor do normal e un incremento da incidencia de cancros.[46] Por exemplo, os ratos deficientes na vía NHEJ (unión de extremos non homólogos) dominante e nos mecanismos de mantemento dos telómeros padecen con maior frecuencia linfomas e infeccións, e, en consecuencia, teñen vidas máis curtas que os ratos de tipo salvaxe.[89] De maneira similar, os ratos deficientes nunha proteína clave de reparación e transcrición que desenrola as hélices de ADN presentan un comezo prematuro de enfermidades relacionadas co envellecemento e un acurtamento da vida.[90] Porén, non todas as deficiencias na reparación do ADN orixinan exactamente os efectos preditos; os ratos deficientes na vía NER mostran un acurtamento da duración da vida sen teren á vez maiores taxas de mutación.[91]
Se a taxa de danos no ADN excede a capacidade da célula de reparala, a acumulación de erros pode causar unha senescencia temperá, apoptose, ou cancro. As doenzas conxénitas asociadas coa reparación defectuosa do ADN causan envellecemento prematuro,[46] o incremento da sensibilidade aos carcinóxenos e o correspondente incremento do risco de cancro (ver máis abaixo). Por outra parte, os organismos con sistemas de reparación do ADN potenciados, como Deinococcus radiodurans, un dos organismos máis resistentes á radiación, mostran unha salientable resistencia aos efectos indutores de roturas de dobre febra da radioactividade, probablemente debido á maior eficiencia da reparación do ADN e especialmente da NHEJ.[92]
Lonxevidade e restrición calórica |
Identificáronse varios xenes que inflúen na variación da duración da vida nunha poboación de organismos. Os efectos destes xenes son moi dependentes do ambiente, en especial, da dieta do organismo. A restrición calórica causa un aumento da duración da vida en varios organismos, probablemente por medio de vías de percepción dos nutrientes e a diminución da taxa metabólica. Os mecanismos moleculares polos cales esta restrición orixina un aumento da vida aínda non están claros (ver S. R. Spindler[93] para unha discusión sobre o asunto); porén, o comportamento de moitos xenes que se sabe que están implicados na reparación do ADN é alterado en condicións de restrición calórica.
Por exemplo, o incremento da dose xénica do xene SIR-2, que regula o empaquetamento do ADN no verme nematodo Caenorhabditis elegans, pode aumentar significativamente a duración da vida.[94] O homólogo en mamíferos de SIR-2 induce factores de reparación do ADN augas abaixo implicados na NHEJ, unha actividade que está promovida especialmente en condicións de restrición calórica.[95] A restrición calórica foi asociada estreitamente coa taxa de reparación por escisión de bases no ADN nuclear de roedores,[96] aínda que non se observaron efectos similares no ADN mitocondrial.[97]
Hai que salientar que o xene AGE-1 de C. elegans, que é un efector augas arriba das vías de reparación do ADN, dá lugar a un drástico aumento da duración da vida en condicións de alimentación libre, pero orixina unha diminución da eficacia reprodutiva en condicións de restrición calórica.[98] Esta observación apoia a teoría da pleiotropía da orixe biolóxica do envellecemento, que propón que os xenes que proporcionan unha ampla vantaxe de supervivencia nas etapas temperás da vida son seleccionados favorablemente aínda que supoñan unha desvantaxe posteriormente nas etapas avanzadas da vida.
Medicina e modulación da reparación do ADN |
Trastornos de reparación do ADN hereditarios |
Os defectos no mecanismo de reparación por escisión de nucleótidos (NER) son responsables de varios trastornos xenéticos, entre os que está os seguintes:[99]
Xeroderma pigmentoso: hipersensibilidade á luz solar/UV, que orixina un aumento da incidencia do cancro de pel e envellecemento prematuro.[100]
Síndrome de Cockayne: hipersensibilidade á luz UV e axentes químicos.[100]
Tricotiodistrofia: pel sensible, pelo e uñas crebadizos.[100]
O atraso mental a miúdo acompaña os dous últimos trastornos, o que suxire un incremento na vulnerabilidade das neuronas do desenvolvemento.
Outros trastornos de reparación do ADN son:
Síndrome de Werner: envellecemento prematuro e retardo no crecemento.[99]
Síndrome de Bloom: hipersensibilidade á luz, alta incidencia de cancros malignos, especialmente leucemias.[101]
Ataxia telanxiectasia: sensibilidade á radiación ionizante e a algúns axentes químicos.[102]
Todos os trastornos mencionados denomínanse con frecuencia "proxerias segmentais" ("enfermidades de envellecemento acelerado") porque as súas vítimas parecen vellas e sofren enfermidades relacionadas co envellecemento a unha idade anormalmente temperá, aínda que non manifestan todos os síntomas da idade avanzada.[103]
Outras enfermidades asociadas cunha función de reparación do ADN reducida son a anemia de Fanconi,[104] o cancro de mama hereditario[105] e o cancro de colon hereditario.[106]
Reparación do ADN e cancro |
A causa de limitacións inherentes aos mecanismos de reparación do ADN, e se as persoas viven o tempo suficiente, finalmente pode dar lugar ao desenvolvemento de cancros.[107][108] Hai polo menos 34 mutacións en xenes de reparación do ADN humanos que incrementan o risco de cancro. Moitas destas mutacións causan que a reparación do ADN sexa menos efectiva do normal. En particular, o cancro colorrectal non poliposo hereditario está fortemente asociado con mutacións específicas na vía de reparación de discordancias no ADN. Os xenes BRCA1 e BRCA2, que son dous famosos xenes cuxas mutacións supoñen un enorme aumento do risco de cancro de mama nos portadores, están ambos os dous asociados cun gran número de vías de reparación do ADN, especialmente a vía NHEJ (unión de extremos non homólogos) e a recombinación homóloga.[109][110]
Os procedementos de terapia do cancro como a quimioterapia e a radioterapia funcionan superando a capacidade da célula de reparar os danos de ADN, o que ten como resultado a morte celular. As células que se ven afectadas preferentemente son as que se dividen máis rapidamente, e as máis típicas son as cancerosas.[111] O efecto colateral é que tamén se ven afectadas outras células non cancerosas pero que tamén se dividen rapidamente como as células proxenitoras do intestino, pel e sistema hematopoético. Os tratamentos modernos do cancro tratan de localizar os danos no ADN só nas células e tecidos asociadas co cancro, xa sexa por medios físicos (concentrando o axente terapéutico na zona do tumor) ou por medios bioquímicos (explotando unha característica única do cancro no corpo).
Defectos de reparación do ADN epixenéticos no cancro |
Clasicamente, o cancro era considerado como un conxunto de enfermidades que se debían a anormalidades xenéticas progresivas entre as que estaban mutacións en xenes supresores de tumores e oncoxenes, e aberracións cromosómicas. Porén, hoxe parece que o cancro tamén se pode orixinar por alteracións epixenéticas.[112]
As alteracións epixenéticas son modificacións relevantes funcionalmente do xenoma que non implican un cambio na secuencia de nucleótidos. Exemplos de ditas modificacións son os cambios na metilación do ADN (hipermetilación e hipometilación), a modificación de histonas,[113] os cambios na arquitectura da cromatina (causada, por exemplo, pola expresión inapropiada de proteínas como HMGA2 ou HMGA1)[114] e os cambios causados polos microARNs. Cada unha destas alteracións epixenéticas serve para regular a expresión xénica sen alterar a secuencia de ADN subxacente. Estes cambios permanecen xeralmente despois da división celular e duran moitas xeracións celulares, polo que poden considerarse como epimutacións (equivalentes a mutacións).
Aínda que nos cancros se encontra un gran número de alteracións epixenéticas, as alteracións epixenéticas en xenes para a reparación do ADN, que causan a expresión reducida de proteínas para a reparación do ADN, parecen ser especialmente importantes. Crese que esas alteracións prodúcense cedo durante o proceso de progresión do cancro e probablemente son a causa da inestabilidade xenética característica dos cancros.[115][116][117][118]
A expresión reducida de xenes para a reparación do ADN causa a reparación deficiente do ADN. Cando a reparación do ADN é deficiente os danos no ADN permanecen nas células a un nivel máis alto do normal e este exceso de danos causa un incremento das frecuencias de mutación ou epimutación. As taxas de mutación increméntanse substancialmente nas células que presentan unha defectuosa reparación de discordancias no ADN[119][120] ou mala reparación recombinacional homóloga (HRR).[121] Os rearranxos cromosómicos e aneuploidías tamén se incrementan nas células con reparación por recombinación homóloga defectuosa.[122]
Os niveis altos de danos no ADN non só causan un incremento das mutacións, senón tamén un incremento da epimutación. Durante a reparación de roturas de dobre febra do ADN, ou a reparación doutros danos no ADN, os sitios de reparación despexados de forma incompleta poden causar un silenciamento de xenes epixenético.[123][124]
A expresión deficiente das proteínas de reparación do ADN debido a unha mutación herdada poden causar un incremento do risco de cancro. Os individuos cunha disfunción herdada en calquera dos 34 xenes de reparación do ADN (trastorno de deficiencia da reparación do ADN) teñen un incremento do risco de cancro, e algúns defectos causan ata un 100% de probabilidade de cancro ao longo da vida (por exemplo, as mutacións p53).[125] Con todo, ditas mutacións na liña xerminal (que causan síndromes cancerosas moi penetrantes) son a causa de só aproximadamente o 1% dos cancros.[126]
Frecuencias de epimutacións en xenes para a reparación do ADN |

Esquema dos axentes que danan o ADN máis comúns, exemplos das lesións que causan no ADN, e vías utilizadas para reparar estas lesións. Tamén se mostran moitos dos xenes destas vías, indícanse cales xenes son regulados epixeneticamente para que teñan unha expresión reducida (ou incrementada) como ocorre en varios cancros. Tamén se mostran os xenes da vía de unión de extremos mediada por microhomoloxía (MMEJ) tendentes ao erro que presentan un incremento da súa expresión en varios cancros.
As deficiencias nos encimas de reparación do ADN orixínanse ocasionalmente por unha mutación somática de nova formación nun xene de reparación do ADN, pero orixínanse moito máis frecuentemente por alteracións epixenéticas que reducen ou silencian a expresión de xenes de reparación do ADN. Por exemplo, cando se examinaron nun estudo 113 cancros colorrectais analizando a súa secuencia, só 4 tiñan unha mutación de cambio de sentido dun codón no xene do encima de reparación do ADN MGMT, aínda que a maioría tiñan unha expresión reducida da MGMT debido á metilación da rexión promotora do xene da MGMT (unha alteración epixenética).[127] En cinco estudos diferentes atopouse que entre o 40% e o 90% dos cancros colorrectais tiñan unha expresión reducida da MGMT debido á metilación da rexión promotora da MGMT.[128][129][130][131][132]
De xeito similar, en 119 casos de cancros colorrectais deficientes na reparación por discordancia na complementariedade que carecían da expresión do xene de reparación do ADN PMS2, atopouse que o PMS2 era deficiente en 6 deles debido a mutacións no xene de PMS2, mentres que noutros 103 casos a expresión de PMS2 era deficiente porque a molécula coa que se asociaba, a MLH1, estaba reprimida debido á metilación do seu promotor (a proteína PMS2 é inestable en ausencia de MLH1).[133] Noutros 10 casos, a perda da expresión de PMS2 era debida probablemente á sobreexpresión epixenética do microARN miR-155, que regula á baixa MLH1.[134]
Noutros casos (tabulados na Táboa 4 desta referencia[135]), encontráronse defectos epixenéticos con frecuencias entre o 13% e o 100% para os xenes de reparación do ADN BRCA1, WRN, FANCB, FANCF, MGMT, MLH1, MSH2, MSH4, ERCC1, XPF, NEIL1 e ATM. Estes defectos epixenéticos aparecían en varios cancros (por exemplo, de mama, ovario, colorrectal e de cabeza e pescozo). Dúas ou tres deficiencias na expresión de ERCC1, XPF ou PMS2 aparecían simultaneamete na maioría dos 49 cancros de colon avaliados por A. Facista et al.[136]
O esquema desta sección mostra algúns axentes que frecuentemente son causantes de danos no ADN, así como exemplos das lesións no ADN que causan, e as vías que teñen que ver con estes danos no ADN. Polo menos existen 169 encimas que se empregan directamente na reparación do ADN ou inflúen nos procesos de reparación do ADN.[137] Deles, hai 83 que se empregan directamente na reparación de 5 tipos de danos no ADN ilustrados no esquema.
No esquema móstranse algúns dos xenes mellor estudados esenciais nestes procesos de reparación. Os xenes escritos en vermello, gris e ciano indican xenes que frecuentemente están alterados epixeneticamente en varios tipos de cancro. Nestes dous artigos resúmese a situación,[135][138] e nestoutros dous artigos sobre dous amplos experimentos[139][140] documentan a maioría das deficiencias na reparación do ADN en cancros.
Os xenes sinalados en vermello son frecuentemente reducidos na súa expresión ou silenciados por mecanismos epixenéticos en varios cancros. Cando estes xenes teñen unha expresión baixa ou ausente, poden acumularse os danos no ADN. A replicación de erros superados estes danos (ver síntese translesión) pode orixinar un incremento das mutacións e, finalmente, o cancro. A represión epixenética dos xenes de reparación do ADN nas vías de reparación do ADN precisas parecen ser fundamentais na carcinoxénese.
Os dous xenes escritos en gris RAD51 e BRCA2, son necesarios para a reparación recombinacional homóloga.[141] Por veces son sobreexpresados epixeneticamente e outras veces subexpresados en certos cancros. Ditos cancros xeralmente teñen deficiencias epixenéticas noutros xenes de reparación do ADN. Estas deficiencias de reparación causan probablemente un incremento de danos no ADN non reparados. A sobreexpresión de RAD51 e BRCA2 observada nestes cancros pode reflectir presións selectivas para a sobreexpresión compensatoria de RAD51 ou BRCA2 e o incremento da reparación recombinacional homóloga para polo menos parcialmente enfrontarse a este exceso de danos. Nestes casos nos que RAD51 ou BRCA2 se subexpresan, isto orixina un incremento de danos no ADN non reparados. A replicación de erros superados estes danos podería causar un incremento de mutacións e de cancro, polo que a subexpresión de RAD51 ou BRCA2 sería en si mesma carcinoxénica.[142]
Os xenes escritos no esquema en cián son da vía unión de extremos mediada por microhomoloxía (MMEJ, do inglés Microhomology-Mediated End Joining) e están regulados á alza no cancro. A MMEJ é unha vía de reparación adicional imprecisa tendente ao erro para as roturas de dobre febra. Nas reparacións MMEJ dunha rotura de dobre febra, unha homoloxía de entre 5 e 25 pares de bases complementarias entre ambas as febras apareadas é suficiente para aliñar as febras, pero xeralmente están presentes extremos con discordancias na complementariedade (solapas). A MMEJ elimina os nucleótidos extra (solapas) onde as febras están unidas, e despois ligan as febras para crear unha dobre hélice do ADN intacta. A MMEJ case sempre implica polo menos unha pequena deleción, polo que é unha vía mutaxénica.[143] A FEN1 é a endonuclease da MMEJ, e é incrementada epixeneticamente por hipometilación do promotor e é sobreexpresada na maioría dos cancros de mama,[144] próstata,[145] de estómago,[146][147] neuroblastomas,[148] de páncreas[149] e de pulmón.[150] A PARP1 tamén se sobreexpresa cando o sitio da súa rexión promotora ETS é hipometilado epixeneticamente, e isto contribúe á progresión do cancro de endometrio,[151] o cancro de ovario co xene BRCA mutado,[152] e o cancro ovárico seroso co BRCA mutado.[153] Outros xenes da vía MMEJ están tamén sobreexpresados en varios cancros, e tamén se mostran no esquema en cor ciano.
En resumo, parece que as alteracións epixenéticas nos xenes de reparación do ADN teñen unha influencia esencial na carcinoxénese.
Reparación do ADN e evolución |
O proceso básico de reparación do ADN está moi conservado tanto entre procariotas coma entre eucariotas e mesmo en virus bacteriófagos (virus que infectan a bacterias); con todo, os organismos máis complexos con xenomas máis complexos teñen, en consecuencia, mecanismos de reparación máis complexos.[154] A capacidade que teñen un gran número de motivos estruturais proteicos de catalizar reaccións químicas relevantes xogou un papel significativo na elaboración de mecanismos de reparación durante a evolución. Pode verse unha revisión extremadamente detallada das hipóteses relacionadas coa evolución da reparación do ADN nesta obra de P. J. O'Brien (2006):[155].
O rexistro fósil indica que a vida unicelular comezou a proliferar no planeta nalgún momento durante o período Precámbrico, aínda que non está claro cando empezou exactamente a primeira forma de vida moderna recoñecible. Os ácidos nucleicos convertéronse nas únicas e universais moléculas que codificaban a información xenética, as cales requirían mecanismos de reparación do ADN, que na súa forma básica foran herdados por todas as formas de vida existentes desde o seu antepasado común. A formación dunha atmosfera rica en oxíxeno na Terra grazas á actividade dos organismos fotosintéticos en determinado momento da súa historia (a coñecida como "catástrofe do oxíxeno") e a presenza de radicais libres potencialmente daniños na célula debido á fosforilación oxidativa fixeron necesaria a evolución de mecanismos de reparación do ADN que actuasen e contrarrestasen especificamente o tipo de danos que produce o estrés oxidativo.[156]
Taxa de cambio evolutivo |
Nalgunhas ocasións, os danos no ADN non son reparados, ou son reparados por medio dun mecanismo que tende a cometer erros, o que ten como resultado un cambio da secuencia orixinal. Cando isto ocorre, as mutacións poden propagarse nos xenomas da proxenie da célula. En caso de que ese evento ocorra nunha célula da liña xerminal que orixine gametos, a mutación ten o potencial de pasar á descendencia do organismo. A taxa de evolución dunha determinada especie (ou dun xene determinado) é unha función da taxa de mutación. En consecuencia, a taxa e a exactitude dos mecanismos de reparación do ADN teñen unha influencia sobre o proceso de cambio evolutivo.[157]
Como a adaptación normal de poboacións de organismos a circunstancias cambiantes (por exemplo a adaptación dos peteiros dunha poboación de pimpíns á presenza cambiante de sementes duras ou insectos) ten lugar por regulación xenética e recombinación e selección de variantes dos xenes (alelos) e non por transmitir á descendencia danos irreparables no ADN,[158] a protección ante os danos no ADN e a súa reparación non inflúe na taxa de adaptación por regulación xénica e por recombinación e selección de alelos. Por outra parte, a reparación dos danos no ADN e a protección si inflúe na taxa de acumulación de mutacións irreparables herdables vantaxosas, que amplían o código, e fai máis lento o mecanismo evolutivo para a expansión do xenoma de organismos con novas funcionalidades. A tensión entre a evolucionabilidade e a reparación das mutacións e protección aínda ten que ser máis investigada.
Notas |
↑ 1,01,1 Lodish H, Berk A, Matsudaira P, Kaiser CA, Krieger M, Scott MP, Zipursky SL, Darnell J. (2004). Molecular Biology of the Cell, p963. WH Freeman: New York, NY. 5th ed.
↑ 2,02,1 Acharya, PV (1971). "The isolation and partial characterization of age-correlated oligo-deoxyribo-ribonucleotides with covalently linked aspartyl-glutamyl polypeptides.". Johns Hopkins medical journal. Supplement (1): 254–60. PMID 5055816.
↑ 3,03,1 Bjorksten, J; Acharya, PV; Ashman, S; Wetlaufer, DB (1971). "Gerogenic fractions in the tritiated rat.". Journal of the American Geriatrics Society 19 (7): 561–74. PMID 5106728. doi:10.1111/j.1532-5415.1971.tb02577.x.
↑ Humpath.com (ed.). "DNA damages". Human pathology. Consultado o 07 de outubro de 2016.
↑ Browner, WS; Kahn, AJ; Ziv, E; Reiner, AP; Oshima, J; Cawthon, RM; Hsueh, WC; Cummings, SR. (2004). "The genetics of human longevity". Am J Med 117 (11): 851–60. PMID 15589490. doi:10.1016/j.amjmed.2004.06.033.
↑ Broad, William J. (7 October 2015). "Nobel Prize in Chemistry Awarded to Tomas Lindahl, Paul Modrich and Aziz Sancar for DNA Studies". New York Times. Consultado o 7 de outubro de 2015.
↑ Staff (7 de outubro de 2015). "THE NOBEL PRIZE IN CHEMISTRY 2015 - DNA repair – providing chemical stability for life" (PDF). Nobel Prize. Consultado o 7 de outubro de 2015.
↑ Jun Guo, Jing Yang, e Yan Li (2015 de xuño de 15). "Association of hOGG1 Ser326Cys polymorphism with susceptibility to hepatocellular carcinoma". Int J Clin Exp Med. 8 (6): 8977–8985. PMC 4537974.(ver sección Discussion)
↑ Encyclopaedia Britannica (ed.). "Tumour suppressor gene". Arquivado dende o orixinal o 07 de abril de 2016. Consultado o 4 de outubro de 2016.
↑ Motoyama, N; Naka, K (febreiro de 2004). "DNA damage tumor suppressor genes and genomic instability". Curr Opin Genet Dev. 14 (1): 11–6. PMID 15108799. doi:10.1016/j.gde.2003.12.003.
↑ Jena, NR (xullo de 2012 Jul). "DNA damage by reactive species: Mechanisms, mutation and repair". Journal of Biosciences 37 (3): 503–17. PMID 22750987.
↑ Povey, Andrew C. (2000). "DNA Adducts: Endogenous and Induced" (PDF). Toxicology and Patology 28 (3): 405–414.
↑ Anthony T. Annunziato. Nature education, ed. "DNA Packaging: Nucleosomes and Chromatin". Scitable. Consultado o 4 de outubro de 2016.
↑ Altaf, Mohammed; Saksouk, Nehmé; Côté, Jacques (1 de maio de 2007). "Histone modifications in response to DNA damage". Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 618 (1–2): 81–90.
↑ 15,015,115,215,315,4 Chakarov, S; Petkova, R; Russev, GCh; Zhelev, N (2014). "DNA damage and mutation. Types of DNA damage.". Biodiscovery 11 (1). doi:10.7750/BioDiscovery.2014.11.1.
↑ 16,016,116,2 Sigma-Aldrich. "Sources of DNA Damage". DNA damage and repair. Consultado o 22-09-2016.
↑ Jackson, Stephen P.; Bartek, Jiri (2009). "The DNA-damage response in human biology and disease". Nature. (461(7267)): 1071–1078. PMC 2906700. doi:10.1038/nature08467.
↑ 18,018,1 Madigan MT, Martino JM (2006). Pearson, ed. Brock Biology of Microorganisms (11th ed.). p. 136. ISBN 0-13-196893-9.
↑ Johns Hopkins Medicina. (ed.). "Cancer Biologists Find DNA-Damaging Toxins in Common Plant-Based Foods. Liquid smoke, black and green teas and coffee produced levels of cell DNA damage comparable to chemo drugs.". News and publications. Consultado o 22-09-2016.
↑ Poirier, Miriam C. (outubro de 2012). "Chemical-induced DNA Damage and Human Cancer Risk". Discovery Medicine.
↑ Roulston A, Marcellus RC, Branton PE (1999). "Viruses and apoptosis". Annu. Rev. Microbiol. 53: 577–628. PMID 10547702. doi:10.1146/annurev.micro.53.1.577.
↑ Ellis, Nathan A.; Ciocci, Susan; German, James (febreiro de 2001). "Back mutation can produce phenotype reversion in Bloom syndrome somatic cells". Human Genetics (Berlin; Nova York: Springer-Verlag) 108 (2): 167–173. ISSN 0340-6717. PMID 11281456. doi:10.1007/s004390000447.
↑ De Bont, Rinne; van Larebeke, Nik (2004). "Endogenous DNA damage in humans: a review of quantitative data.". Mutagenesis (Oxford University Press) 19 (3): 169–185. doi:10.1093/mutage/geh025.
↑ Cooke, Marcus S.; Evans, Mark D.; Dizdaroglu, Miral; Lunec, Joseph (xullode 2003). "Oxidative DNA damage: mechanisms, mutation, and disease". The FASEB Journal 17 (10): 1195–1214. doi:10.1096/fj.02-0752rev.
↑ Baylln, S. B.; Herman, J. G.; Graff, J. R.; Vertino, P. M.; Issa, J. P. (1997). "Alterations in DNA Methylation: A Fundamental Aspect of Neoplasia". Advances in Cancer Research Volume 72. Advances in Cancer Research 72. p. 141. doi 10.1016/S0065-230X(08)60702-2. ISBN 978-0-12-006672-8.
↑ Robert Shapiro. Damage to DNA Caused by Hydrolysis (volume 40 of the series NATO Advanced Study Institutes Series). Springler. pp. 3–18.
↑ Siede, Wolfram; Doetsch, Paul W. DNA Damage Recognition. Google books. pp. 265–266.
↑ 28,028,1 Kendric Smith. Photochemical and Photobiological Reviews. Google books. Páxinas 274-275.
↑ M. J. Peak, J. G. Peak. Effects of Solar Ultraviolet Photons on Mammalian Cell DNA
↑ Hanson Kerry M.; Gratton Enrico; Bardeen Christopher J. (2006). "Sunscreen enhancement of UV-induced reactive oxygen species in the skin". Free Radical Biology and Medicine 41 (8): 1205–1212. PMID 17015167. doi:10.1016/j.freeradbiomed.2006.06.011.
↑ Ward JF. (1988). "DNA damage produced by ionizing radiation in mammalian cells: identities, mechanisms of formation, and reparability.". Prog Nucleic Acid Res Mol Biol. (35): 95–125. PMID 3065826.
↑ Ohta, Toshihiro; Shin-ichi, Tokishita; Mochizuki, Kayo; Kawase, Jun; Sakahira, Masahide; Yamagata, Hideo (2006). "UV Sensitivity and Mutagenesis of the Extremely Thermophilic Eubacterium Thermus thermophilus HB27". Genes and Environment 28 (2): 56–61. doi:10.3123/jemsge.28.56.
↑ B. Singer (febreiro de 1996). "DNA Damage: Chemistry, Repair, and Mutagenic Potential". Regulatory Toxicology and Pharmacology (Elsevier) 23 (1): 2–13. doi:10.1006/rtph.1996.0002.
↑ Driessens, N; Versteyhe, S; Ghaddhab, C; Burniat, A; De Deken, X; Van Sande, J; Dumont, JE; Miot, F; Corvilain, B (setembro de 2009). "Hydrogen peroxide induces DNA single- and double-strand breaks in thyroid cells and is therefore a potential mutagen for this organ.". Endocr Relat Cancer 16 (3): 845–56. PMID 19509065. doi:10.1677/ERC-09-0020.
↑ Chen, SY (2002). "Polycyclic Aromatic Hydrocarbon-DNA Adducts in Liver Tissues of Hepatocellular Carcinoma Patients and Controls". Int J Cancer (99): 14–21. PMID 11948486.
↑ Baird, W. M.; Hooven, L. A.; Mahadevan, B. "Carcinogenic polycyclic aromatic hydrocarbon-DNA adducts and mechanism of action". Environmental and Molecular Mutagenesis 45 (2-3): 106–114. ISSN 1098-2280. doi:10.1002/em.20095.
↑ Eissenberg, Joel C. (2002). Gale Group Inc., ed. "Eukaryotic chromosome". Encyclopedia.com. Consultado o 3 de outubro de 2016.
↑ Blasiak, J; Hoser, G; Bialkowska-Warzecha, J; Pawlowska, E; Skorski, T (2015). "Reactive Oxygen Species and Mitochondrial DNA Damage and Repair in BCR-ABL1 Cells Resistant to Imatinib.". Biores Open Access. 4 (1): 334–42. PMID 26309809.
↑ Shokolenko, Inna; Venediktova, Natalia; Bochkareva, Alexandra; Wilson, Glenn L.; Alexeyev, Mikhail F (2009). "Oxidative stress induces degradation of mitochondrial DNA.". Nucleic Acids Res. 37 (8): 2539–2548. PMC 2677867. doi:10.1093/nar/gkp100.
↑ Ishikawa, Kaori; Takenaga, Keizo; Akimoto, Miho; Koshikawa, Nobuko; Yamaguchi, Aya; Imanishi, Hirotake; Nakada, Kazuto; Honma, Yoshio; Hayashi, Jun-Ichi (maio 2008). "ROS-Generating Mitochondrial DNA Mutations Can Regulate Tumor Cell Metastasis". Science 320 (5876).
↑ Braig, M; Schmitt, CA. (2006). "Oncogene-induced senescence: putting the brakes on tumor development". Cancer Res 66 (6): 2881–2884. PMID 16540631. doi:10.1158/0008-5472.CAN-05-4006.
↑ Lynch, MD. (2006). "How does cellular senescence prevent cancer?". DNA Cell Biol 25 (2): 69–78. PMID 16460230. doi:10.1089/dna.2006.25.69.
↑ Campisi J, d'Adda di Fagagna F (2007). "Cellular senescence: when bad things happen to good cells.". Rev Mol Cell Biol. 8 (9): 729–40. PMID 17667954. doi:10.1038/nrm2233.
↑ "DNA damage". Glosario do NCBI. Consultado o 30 de setembro de 2016.
↑ "Mutation". Glosartio do NCBI. Consultado o 30 de setembro de 2016.
↑ 46,046,146,2 Best, Benjamin P (2009). "Nuclear DNA damage as a direct cause of aging" (PDF). Rejuvenation Research 12 (3): 199–208. PMID 19594328. doi:10.1089/rej.2009.0847.
↑ Geoffrey M Cooper. (2000). "DNA repair (on line)". The Cell: A Molecular Approach (2nd. ed.). Sinauer Associates. ISBN 0-87893-106-6.
↑ 48,048,1 T. S. Dexheimer. DNA Repair Pathways and Mechanisms. Springer.
↑ Sancar, A. (2003). "Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors". Chem Rev 103 (6): 2203–37. PMID 12797829. doi:10.1021/cr0204348.
↑ José Ignacio Lucas-Lledó, Michael Lynch; Lynch, M. (19 de febreiro de 2009). "Evolution of Mutation Rates: Phylogenomic Analysis of the Photolyase/Cryptochrome Family". Molecular Biology and Evolution 26 (5): 1143–1153. PMC 2668831. PMID 19228922. doi:10.1093/molbev/msp029.
↑ 51,051,151,2 Watson JD, Baker TA, Bell SP, Gann A, Levine M, Losick R. (2004). Molecular Biology of the Gene. Pearson Benjamin Cummings; CSHL Press. 5th ed., chapters 9 and 10
↑ Volkert MR (1988). "Adaptive response of Escherichia coli to alkylation damage". Environmental and Molecular Mutagenesis 11 (2): 241–55. PMID 3278898. doi:10.1002/em.2850110210.
↑ 53,053,153,2 Willey, J; Sherwood, L; Woolverton, C (2014). McGraw Hill, ed. Prescott’s Microbiology. New York, New York. p. 381. ISBN 978-0-07-3402-40-6.
↑ 54,054,154,254,3 Reardon, J; Sancar, A (2006). "Purification and Characterization of Escherichia coli and Human Nucleotide Excision Repair Enzyme Systems". Methods in Enzymology 408: 189–213. PMID 16793370. doi:10.1016/S0076-6879(06)08012-8.
↑ Berg, M; Tymoczko, J; Stryer, L (2012). W.H. Freeman and Company, ed. Biochemistry 7th edition. New York. p. 840. ISBN 9781429229364.
↑ Wilson TE, Grawunder U, Lieber MR (July 1997). "Yeast DNA ligase IV mediates non-homologous DNA end joining". Nature 388 (6641): 495–8. PMID 9242411. doi:10.1038/41365.
↑ Moore JK, Haber JE (maio de 1996). "Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae". Molecular and Cellular Biology 16 (5): 2164–73. PMC 231204. PMID 8628283.
↑ Boulton SJ, Jackson SP (setembro de 1996). "Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways". The EMBO Journal 15 (18): 5093–103. PMC 452249. PMID 8890183.
↑ Wilson TE, Lieber MR (agosto de 1999). "Efficient processing of DNA ends during yeast nonhomologous end joining. Evidence for a DNA polymerase beta (Pol4)-dependent pathway". The Journal of Biological Chemistry 274 (33): 23599–609. PMID 10438542. doi:10.1074/jbc.274.33.23599.
↑ Budman J, Chu G (febreiro de 2005). "Processing of DNA for nonhomologous end-joining by cell-free extract". The EMBO Journal 24 (4): 849–60. PMC 549622. PMID 15692565. doi:10.1038/sj.emboj.7600563.
↑ Wang H, Perrault AR, Takeda Y, Qin W, Wang H, Iliakis G (setembro de 2003). "Biochemical evidence for Ku-independent backup pathways of NHEJ". Nucleic Acids Research 31 (18): 5377–88. PMC 203313. PMID 12954774. doi:10.1093/nar/gkg728.
↑ Jung D, Alt FW (xaneiro de 2004). "Unraveling V(D)J recombination; insights into gene regulation". Cell 116 (2): 299–311. PMID 14744439. doi:10.1016/S0092-8674(04)00039-X.
↑ 63,063,1 Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, Razavian N, Berns MW, Wu X (2013). "Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells". Proc. Natl. Acad. Sci. U.S.A. 110 (19): 7720–5. PMC 3651503. PMID 23610439. doi:10.1073/pnas.1213431110.
↑ Sharma S, Javadekar SM, Pandey M, Srivastava M, Kumari R, Raghavan SC (2015). "Homology and enzymatic requirements of microhomology-dependent alternative end joining". Cell Death Dis 6: e1697. PMC 4385936. PMID 25789972. doi:10.1038/cddis.2015.58.
↑ Liang L, Deng L, Chen Y, Li GC, Shao C, Tischfield JA (2005). "Modulation of DNA end joining by nuclear proteins". J. Biol. Chem. 280 (36): 31442–9. PMID 16012167. doi:10.1074/jbc.M503776200.
↑ Decottignies A (2013). "Alternative end-joining mechanisms: a historical perspective". Front Genet 4: 48. PMC 3613618. PMID 23565119. doi:10.3389/fgene.2013.00048.
↑ Jasin M, Rothstein R (novembro de 2013). "Repair of strand breaks by homologous recombination". Cold Spring Harbor Perspectives in Biology 5 (11): a012740. PMC 3809576. PMID 24097900. doi:10.1101/cshperspect.a012740.
↑ Resnick MA (xuño de 1976). "The repair of double-strand breaks in DNA; a model involving recombination". Journal of Theoretical Biology 59 (1): 97–106. PMID 940351. doi:10.1016/s0022-5193(76)80025-2.
↑ Champoux JJ (2001). "DNA topoisomerases: structure, function, and mechanism". Annu. Rev. Biochem. 70: 369–413. PMID 11395412. doi:10.1146/annurev.biochem.70.1.369.
↑ Zahradka K, Slade D, Bailone A; et al. (outubro de 2006). "Reassembly of shattered chromosomes in Deinococcus radiodurans". Nature 443 (7111): 569–73. PMID 17006450. doi:10.1038/nature05160.
↑ Waters LS, Minesinger BK, Wiltrout ME, D'Souza S, Woodruff RV, Walker GC (marzo de 2009). "Eukaryotic Translesion Polymerases and Their Roles and Regulation in DNA Damage Tolerance". Microbiol. Mol. Biol. Rev. 73 (1): 134–54. PMC 2650891. PMID 19258535. doi:10.1128/MMBR.00034-08.
↑ Julian E. Sale (2013). "Translesion DNA Synthesis and Mutagenesis in Eukaryotes". Cold Spring Harbor Laboratory Press. doi:10.1101/cshperspect.a012708.
↑ 73,073,1 Prakash, Satya; Prakash, Louise (2002). "Translesion DNA synthesis in eukaryotes: A one- or two-polymerase affair". Genes & Developement (Cold Spring Harbor Laboratory Press) (16): 1872–1883. doi:10.1101/gad.1009802.
↑ LC Colis, P Raychaudhury, AK Basu (2008). "Mutational specificity of gamma-radiation-induced guanine-thymine and thymine-guanine intrastrand cross-links in mammalian cells and translesion synthesis past the guanine-thymine lesion by human DNA polymerase eta". Biochemistry 47 (6): 8070–8079. PMID 18616294. doi:10.1021/bi800529f.
↑ P Raychaudhury; Basu, Ashis K. (2011). "Genetic requirement for mutagenesis of the G[8,5-Me]T cross-link in Escherichia coli: DNA polymerases IV and V compete for error-prone bypass". Biochemistry 50 (12): 2330–2338. PMID 21302943. doi:10.1021/bi102064z.
↑ PennState. Benkovich Laboratory (ed.). "Translesion DNA Synthesis". Pennsylvania State University (PennState). Consultado o 24-09-2016.
↑ Wang, Z (2001). "Translesion synthesis by the UmuC family of DNA polymerases". Mutat. Res. 486 (2): 59–70. PMID 11425512. doi:10.1016/S0921-8777(01)00089-1.
↑ 78,078,178,2 Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. (2006). ASM Press., ed. DNA Repair and Mutagenesis, part 3. (2nd ed.).
↑ Vermeulen, Katrien; Van Bockstaele, Dirk R.; Berneman, Zwi N. (June 2003). "The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer". Cell Proliferation 36 (3): 131–149. doi:10.1046/j.1365-2184.2003.00266.x.
↑ Bakkenist CJ, Kastan MB (xaneiro de 2003). "DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation". Nature 421 (6922): 499–506. PMID 12556884. doi:10.1038/nature01368.
↑ Wei, Qingyi; Lei Li; David Chen (2007). World Scientific, ed. DNA Repair, Genetic Instability, and Cancer. ISBN 981-270-014-5.
↑ Shiloh, Y; Kastan, MB (2001). "ATM: genome stability, neuronal development, and cancer cross paths.". Advances in cancer research 83: 209–54. PMID 11665719. doi:10.1016/s0065-230x(01)83007-4.
↑ Schonthal, Axel H. (2004). Humana Press, ed. Checkpoint Controls and Cancer. ISBN 1-58829-500-1.
↑ Gartel AL, Tyner AL (xuño de 2002). "The role of the cyclin-dependent kinase inhibitor p21 in apoptosis". Molecular Cancer Therapeutics 1 (8): 639–49. PMID 12479224.
↑ Janion, C. (2001). "Some aspects of the SOS response system-a critical survey". Acta Biochim Pol. 48 (3): 599–610. PMID 11833768.
↑ 86,086,1 Erill, I; Campoy, S; Barbé, J (2007). "Aeons of distress: an evolutionary perspective on the bacterial SOS response". FEMS Microbiol Rev. 31 (6): 637–656. PMID 17883408. doi:10.1111/j.1574-6976.2007.00082.x.
↑ Schlacher, K; Pham, P; Cox, MM; Goodman, MF. (2006). "Roles of DNA Polymerase V and RecA Protein in SOS Damage-Induced Mutation". Chem. Rev 106 (2): 406–419. PMID 16464012. doi:10.1021/cr0404951.
↑ Fry, RC; Begley, TJ; Samson, LD (2004). "Genome-wide responses to DNA-damaging agents". Annu Rev Microbiol 59: 357–77. PMID 16153173. doi:10.1146/annurev.micro.59.031805.133658.
↑ Espejel, S; Martin, M; Klatt, P; Martin-Caballero, J; Flores, JM; Blasco, MA. (2004). "Shorter telomeres, accelerated ageing and increased lymphoma in DNA-PKcs-deficient mice". EMBO Rep 5 (5): 503–9. PMC 1299048. PMID 15105825. doi:10.1038/sj.embor.7400127.
↑ De Boer, J; Andressoo, JO; De Wit, J; Huijmans, J; Beems, RB; Van Steeg, H; Weeda, G; Van Der Horst, GT; et al. (2002). "Premature aging in mice deficient in DNA repair and transcription". Science 296 (5571): 1276–9. PMID 11950998. doi:10.1126/science.1070174.
↑ Dolle, ME; Busuttil, RA; Garcia, AM; Wijnhoven, S; van Drunen, E; Niedernhofer, LJ; der Horst, G; Hoeijmakers, JH; et al. (2006). "Increased genomic instability is not a prerequisite for shortened life span in DNA repair deficient mice". Mutation Research 596 (1–2): 22–35. PMID 16472827. doi:10.1016/j.mrfmmm.2005.11.008.
↑ Kobayashi, Y; Narumi, I; Satoh, K; Funayama, T; Kikuchi, M; Kitayama, S; Watanabe, H. (2004). "Radiation response mechanisms of the extremely radioresistant bacterium Deinococcus radiodurans". Biol Sci Space 18 (3): 134–5. PMID 15858357.
↑ Spindler, SR. (2005). "Rapid and reversible induction of the longevity, anticancer and genomic effects of caloric restriction". Mech Ageing Dev 126 (9): 960–6. PMID 15927235. doi:10.1016/j.mad.2005.03.016.
↑ Tissenbaum, HA; Guarente, L. (2001). "Increased dosage of a sir-2 gene extends life span in Caenorhabditis elegans". Nature 410 (6825): 227–30. PMID 11242085. doi:10.1038/35065638.
↑ Cohen, HY; Miller, C; Bitterman, KJ; Wall, NR; Hekking, B; Kessler, B; Howitz, KT; Gorospe, M; et al. (2004). "Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase". Science 305 (5682): 390–2. PMID 15205477. doi:10.1126/science.1099196.
↑ Cabelof, DC; Yanamadala, S; Raffoul, JJ; Guo, Z; Soofi, A; Heydari, AR. (2003). "Caloric restriction promotes genomic stability by induction of base excision repair and reversal of its age-related decline". DNA Repair (Amst.) 2 (3): 295–307. PMID 12547392. doi:10.1016/S1568-7864(02)00219-7.
↑ Stuart, JA; Karahalil, B; Hogue, BA; Souza-Pinto, NC; Bohr, VA. (2004). "Mitochondrial and nuclear DNA base excision repair are affected differently by caloric restriction". FASEB J 18 (3): 595–7. PMID 14734635. doi:10.1096/fj.03-0890fje.
↑ Walker, DW; McColl, G; Jenkins, NL; Harris, J; Lithgow, GJ. (2000). "Evolution of lifespan in C. elegans". Nature 405 (6784): 296–7. PMID 10830948. doi:10.1038/35012693.
↑ 100,0100,1100,2 de Boer, Jan; Hoeijmakers, Jan H.J. (2000). "Nucleotide excision repair and human syndromes". Carcinogenesis (Oxford Journals) 3 (21): 453–460. doi:10.1093/carcin/21.3.453.
↑ Kristian Moss Bendtsen; Martin Borch Jensen; Alfred May; Lene Juel Rasmussen; Ala Trusina; Vilhelm A. Bohr; Mogens H. Jensen (2014). "Dynamics of the DNA repair proteins WRN and BLM in the nucleoplasm and nucleoli". European Biophysics Journal 43: 509–16. PMID 25119658. doi:10.1007/s00249-014-0981-x.
↑ Colton, SL; Xu, XS; Wang, YA; Wang, G. (setembro de 2006). "The involvement of ataxia-telangiectasia mutated protein activation in nucleotide excision repair-facilitated cell survival with cisplatin treatment.". J Biol Chem. 37 (281): 27117–25. PMID 16849332.
↑ Christopher, A. E.; Dyer, Alan; Sinclair, N J. (1998). "The premature ageing syndromes:insights into the ageing process" (PDF). Age and Ageing (27): 73–80.Existen varias condicións raras no home que mostran certas características fenotípicas asociadas coa senescencia. A miúdo denomínanse 'síndromes proxeroides segmentais', os máis amplamente estudados deles son a proxeria de Hutchinson-Gilford, a síndrome de Werner e a síndrome de Cokayne, pero o grupo tamén inclúe a síndrome de Bloom, a ataxia telanxiectasia e a sínrome de Down (Páxina 73). Orixinal:Several rare conditions exist in man that exhibit certain phenotypic characteristics associated with senescence. Often referred to as 'segmental progeroid syndromes', the most widely studied of these are Hutchinson-Gilford progeria, Werner's syndrome and Cockayne's syndrome, but the group also includes Bloom's syndrome, ataxia telangiectasia and Down's syndrome. (páxina 73).
↑ Mouw, KW; D'Andrea, AD (xullo de 2014). "Crosstalk between the nucleotide excision repair and Fanconi anemia/BRCA pathways.". DNA Repair (Amst). 130-4 (19). PMID 24768451. doi:10.1016/j.dnarep.2014.03.019. .
↑ Jean J. Latimera, Jennifer M. Johnsonb, Crystal M. Kellya, Tiffany D. Milesb, Kelly A. Beaudry-Rodgersd, Nancy A. Lalanneb, Victor G. Vogelb, Amal Kanbour-Shakirf, Joseph L. Kelleya, Ronald R. Johnsong, and Stephen G. Grant. PNAS, ed. "Nucleotide excision repair deficiency is intrinsic in sporadic stage I breast cancer" 107 (50): 21725–21730. doi:10.1073/pnas.0914772107.
↑ Berndt, SI; Platz, EA; Fallin, MD; Thuita, LW; Hoffman, SC; Helzlsouer, KJ (novembro de 2006). "Genetic variation in the nucleotide excision repair pathway and colorectal cancer risk.". Cancer Epidemiol Biomarkers Prev. 11 (15): 2263–9. PMID 17119055. doi:10.1158/1055-9965.EPI-06-0449.
↑ Johnson, George (28 December 2010). "Unearthing Prehistoric Tumors, and Debate". The New York Times.Se vivimos o suficiente, máis cedo ou máis tarde teremos cancro.
↑ Alberts, B, Johnson A, Lewis J; et al. (2002). "The Preventable Causes of Cancer". En Garland Science. Molecular biology of the cell (4th ed.). New York. ISBN 0-8153-4072-9.Unha certa incidencia de fondo irreducible de cancro é esperable sen importar as circunstancias: as mutacións nunca poden ser absolutamente evitadas, porque son unha inevitable consecuencia das limitacións fundamentais na replicación do ADN precisa, como se discute no capítulo 5. Se unha persoa viven o suficiente, é inevitable que polo menos unha das súas células acumularía finalmente un conxunto de mutacións que seria dabondo para desenvolver un cancro.
↑ Bau, DT; Mau, YC; Shen, CY (agosto de 2006). "The role of BRCA1 in non-homologous end-joining". Cancer Lett (240 1volume=1): 1–8. PMID 16171943. doi:10.1016/j.canlet.2005.08.003.
↑ Fen Xia, Danielle G. Taghian, Jeffrey S. DeFrank, Zhao-Chong Zeng, Henning Willers, George Iliakis, and Simon N. Powell. "Deficiency of human BRCA2 leads to impaired homologous recombination but maintains normal nonhomologous end joining" 98 (15): 8644–8649. doi:10.1073/pnas.151253498.
↑ Siddik ZH (2005). John Wiley & Sons, Ltd, ed. Mechanisms of Action of Cancer Chemotherapeutic Agents: DNA-Interactive Alkylating Agents and Antitumour Platinum-Based Drugs. doi:10.1002/0470025077.chap84b.
↑ Baylin SB; Ohm, Joyce E. (febreiro de 2006). "Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction?". Nat. Rev. Cancer 6 (2): 107–16. PMID 16491070. doi:10.1038/nrc1799.
↑ Kanwal R, Gupta S, 4 (abril de 2012). "Epigenetic modifications in cancer". Clinical Genetics 81: 303–11. PMC 3590802. PMID 22082348. doi:10.1111/j.1399-0004.2011.01809.x.
↑ Baldassarre G, Battista S, Belletti B; et al. (abril de 2003). "Negative regulation of BRCA1 gene expression by HMGA1 proteins accounts for the reduced BRCA1 protein levels in sporadic breast carcinoma". Molecular and Cellular Biology 23 (7): 2225–38. PMC 150734. PMID 12640109. doi:10.1128/MCB.23.7.2225-2238.2003.
↑ Jacinto FV, Esteller M; Esteller, M. (xullo de 2007). "Mutator pathways unleashed by epigenetic silencing in human cancer". Mutagenesis 22 (4): 247–53. PMID 17412712. doi:10.1093/mutage/gem009.
↑ Lahtz C; Pfeifer, G. P. (febreiro de 2011). "Epigenetic changes of DNA repair genes in cancer". J Mol Cell Biol 3 (1): 51–8. PMC 3030973. PMID 21278452. doi:10.1093/jmcb/mjq053. http://jmcb.oxfordjournals.org/content/3/1/51.long
↑ Bernstein C, Nfonsam V, Prasad AR, Bernstein H (marzo de 2013). "Epigenetic field defects in progression to cancer". World J Gastrointest Oncol 5 (3): 43–9. PMC 3648662. PMID 23671730. doi:10.4251/wjgo.v5.i3.43.
↑ Bernstein C, Prasad AR, Nfonsam V, Bernstein H. (2013). Prof. Clark Chen, ed. New Research Directions in DNA Repair. Chapter 16: DNA Damage, DNA Repair and Cancer. InTech. ISBN 978-953-511-114-6.
↑ Narayanan L, Fritzell JA, Baker SM, Liskay RM, Glazer PM (abril de 1997). "Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2". Proceedings of the National Academy of Sciences of the United States of America 94 (7): 3122–7. PMC 20332. PMID 9096356. doi:10.1073/pnas.94.7.3122.
↑ Hegan DC, Narayanan L, Jirik FR, Edelmann W, Liskay RM, Glazer PM (decembro de 2006). "Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6". Carcinogenesis 27 (12): 2402–8. PMC 2612936. PMID 16728433. doi:10.1093/carcin/bgl079.
↑ Tutt AN, van Oostrom CT, Ross GM, van Steeg H, Ashworth A (marzo de 2002). "Disruption of Brca2 increases the spontaneous mutation rate in vivo: synergism with ionizing radiation". EMBO Reports 3 (3): 255–60. PMC 1084010. PMID 11850397. doi:10.1093/embo-reports/kvf037.
↑ German J (marzo de 1969). "Bloom's syndrome. I. Genetical and clinical observations in the first twenty-seven patients". American Journal of Human Genetics 21 (2): 196–227. PMC 1706430. PMID 5770175.
↑ O'Hagan HM, Mohammad HP, Baylin SB (2008). "Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island". PLoS Genetics 4 (8): e1000155. PMC 2491723. PMID 18704159. doi:10.1371/journal.pgen.1000155.
↑ Cuozzo C, Porcellini A, Angrisano T; et al. (xullo de 2007). "DNA damage, homology-directed repair, and DNA methylation". PLoS Genetics 3 (7): e110. PMC 1913100. PMID 17616978. doi:10.1371/journal.pgen.0030110.
↑ Malkin D (abril de 2011). "Li-fraumeni syndrome". Genes & Cancer 2 (4): 475–84. PMC 3135649. PMID 21779515. doi:10.1177/1947601911413466.
↑ Fearon ER (novembro de 1997). "Human cancer syndromes: clues to the origin and nature of cancer". Science 278 (5340): 1043–50. PMID 9353177. doi:10.1126/science.278.5340.1043.
↑ Halford S, Rowan A, Sawyer E, Talbot I, Tomlinson I (xuñp de 2005). "O(6)-methylguanine methyltransferase in colorectal cancers: detection of mutations, loss of expression, and weak association with G:C>A:T transitions". Gut 54 (6): 797–802. PMC 1774551. PMID 15888787. doi:10.1136/gut.2004.059535.
↑ Shen L, Kondo Y, Rosner GL; et al. (setembro de 2005). "MGMT promoter methylation and field defect in sporadic colorectal cancer". Journal of the National Cancer Institute 97 (18): 1330–8. PMID 16174854. doi:10.1093/jnci/dji275.
↑ Psofaki V, Kalogera C, Tzambouras N; et al. (xullo de 2010). "Promoter methylation status of hMLH1, MGMT, and CDKN2A/p16 in colorectal adenomas". World Journal of Gastroenterology 16 (28): 3553–60. PMC 2909555. PMID 20653064. doi:10.3748/wjg.v16.i28.3553.
↑ Lee KH, Lee JS, Nam JH; et al. (outubro de 2011). "Promoter methylation status of hMLH1, hMSH2, and MGMT genes in colorectal cancer associated with adenoma-carcinoma sequence". Langenbeck's Archives of Surgery 396 (7): 1017–26. PMID 21706233. doi:10.1007/s00423-011-0812-9.
↑ Amatu A, Sartore-Bianchi A, Moutinho C; et al. (abril de 2013). "Promoter CpG island hypermethylation of the DNA repair enzyme MGMT predicts clinical response to dacarbazine in a phase II study for metastatic colorectal cancer". Clinical Cancer Research 19 (8): 2265–72. PMID 23422094. doi:10.1158/1078-0432.CCR-12-3518.
↑ Mokarram P, Zamani M, Kavousipour S; et al. (maio de 2013). "Different patterns of DNA methylation of the two distinct O6-methylguanine-DNA methyltransferase (O6-MGMT) promoter regions in colorectal cancer". Molecular Biology Reports 40 (5): 3851–7. PMID 23271133. doi:10.1007/s11033-012-2465-3.
↑ Truninger K, Menigatti M, Luz J; et al. (maio de 2005). "Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer". Gastroenterology 128 (5): 1160–71. PMID 15887099. doi:10.1053/j.gastro.2005.01.056.
↑ Valeri N, Gasparini P, Fabbri M; et al. (abril de 2010). "Modulation of mismatch repair and genomic stability by miR-155". Proceedings of the National Academy of Sciences of the United States of America 107 (15): 6982–7. PMC 2872463. PMID 20351277. doi:10.1073/pnas.1002472107.
↑ 135,0135,1 Carol Bernstein and Harris Bernstein (2015). Epigenetic Reduction of DNA Repair in Progression to Cancer, Advances in DNA Repair, Prof. Clark Chen (Ed.), ISBN 978-953-512-209-8, InTech, Available from: http://www.intechopen.com/books/advances-in-dna-repair/epigenetic-reduction-of-dna-repair-in-progression-to-cancer
↑ Facista A, Nguyen H, Lewis C; et al. (2012). "Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer". Genome Integrity 3 (1): 3. PMC 3351028. PMID 22494821. doi:10.1186/2041-9414-3-3.
↑ MD Anderson Cancer Center, University of Texas, ed. (15 de abril de 2014). "Human DNA Repair Genes".
↑ Xinrong Chen and Tao Chen (2011. url=http://www.intechopen.com/books/dnarepair/roles-of-microrna-in-dna-damage-and-repair Chapter 18: Roles of MicroRNA in DNA Damage and Repair). Inna Kruman, ed. DNA Repair. InTech. ISBN 978-953-307-697-3.
↑ Krishnan K, Steptoe AL, Martin HC (febreiro de 2013). "MicroRNA-182-5p targets a network of genes involved in DNA repair". RNA 19 (2): 230–42. PMC 3543090. PMID 23249749. doi:10.1261/rna.034926.112.
↑ Chaisaingmongkol J, Popanda O, Warta R (decembro de 2012). "Epigenetic screen of human DNA repair genes identifies aberrant promoter methylation of NEIL1 in head and neck squamous cell carcinoma". Oncogene 31 (49): 5108–16. PMID 22286769. doi:10.1038/onc.2011.660.
↑ Qingyi Wei, Dr. Lei Li, Dr. David Chen. DNA Repair, Genetic Instability, and Cancer. Google books. Páxina 135-136
↑ Klein HL (2008). "The consequences of Rad51 overexpression for normal and tumor cells". DNA Repair (Amst.) 7 (5): 686–93. PMC 2430071. PMID 18243065. doi:10.1016/j.dnarep.2007.12.008.
↑ Liang L, Deng L, Chen Y, Li GC, Shao C, Tischfield JA (2005). "Modulation of DNA end joining by nuclear proteins". J. Biol. Chem. 280 (36): 31442–9. PMID 16012167. doi:10.1074/jbc.M503776200.
↑ Singh P, Yang M, Dai H, Yu D, Huang Q, Tan W, Kernstine KH, Lin D, Shen B (2008). "Overexpression and hypomethylation of flap endonuclease 1 gene in breast and other cancers". Mol. Cancer Res. 6 (11): 1710–7. PMC 2948671. PMID 19010819. doi:10.1158/1541-7786.MCR-08-0269.
↑ Lam JS, Seligson DB, Yu H, Li A, Eeva M, Pantuck AJ, Zeng G, Horvath S, Belldegrun AS (2006). "Flap endonuclease 1 is overexpressed in prostate cancer and is associated with a high Gleason score". BJU Int. 98 (2): 445–51. PMID 16879693. doi:10.1111/j.1464-410X.2006.06224.x.
↑ Kim JM, Sohn HY, Yoon SY, Oh JH, Yang JO, Kim JH, Song KS, Rho SM, Yoo HS, Yoo HS, Kim YS, Kim JG, Kim NS (2005). "Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells". Clin. Cancer Res. 11 (2 Pt 1): 473–82. PMID 15701830.
↑ Wang K, Xie C, Chen D (2014). "Flap endonuclease 1 is a promising candidate biomarker in gastric cancer and is involved in cell proliferation and apoptosis". Int. J. Mol. Med. 33 (5): 1268–74. PMID 24590400. doi:10.3892/ijmm.2014.1682.
↑ Krause A, Combaret V, Iacono I, Lacroix B, Compagnon C, Bergeron C, Valsesia-Wittmann S, Leissner P, Mougin B, Puisieux A (2005). "Genome-wide analysis of gene expression in neuroblastomas detected by mass screening". Cancer Lett. 225 (1): 111–20. PMID 15922863. doi:10.1016/j.canlet.2004.10.035.
↑ Iacobuzio-Donahue CA, Maitra A, Olsen M, Lowe AW, van Heek NT, Rosty C, Walter K, Sato N, Parker A, Ashfaq R, Jaffee E, Ryu B, Jones J, Eshleman JR, Yeo CJ, Cameron JL, Kern SE, Hruban RH, Brown PO, Goggins M (2003). "Exploration of global gene expression patterns in pancreatic adenocarcinoma using cDNA microarrays". Am. J. Pathol. 162 (4): 1151–62. PMC 1851213. PMID 12651607. doi:10.1016/S0002-9440(10)63911-9.
↑ Sato M, Girard L, Sekine I, Sunaga N, Ramirez RD, Kamibayashi C, Minna JD (2003). "Increased expression and no mutation of the Flap endonuclease (FEN1) gene in human lung cancer". Oncogene 22 (46): 7243–6. PMID 14562054. doi:10.1038/sj.onc.1206977.
↑ Bi FF, Li D, Yang Q (2013). "Hypomethylation of ETS transcription factor binding sites and upregulation of PARP1 expression in endometrial cancer". Biomed Res Int 2013: 946268. PMC 3666359. PMID 23762867. doi:10.1155/2013/946268.
↑ Li D, Bi FF, Cao JM, Cao C, Li CY, Liu B, Yang Q (2014). "Poly (ADP-ribose) polymerase 1 transcriptional regulation: a novel crosstalk between histone modification H3K9ac and ETS1 motif hypomethylation in BRCA1-mutated ovarian cancer". Oncotarget 5 (1): 291–7. PMC 3960209. PMID 24448423. doi:10.18632/oncotarget.1549.
↑ Bi FF, Li D, Yang Q (2013). "Promoter hypomethylation, especially around the E26 transformation-specific motif, and increased expression of poly (ADP-ribose) polymerase 1 in BRCA-mutated serous ovarian cancer". BMC Cancer 13: 90. PMC 3599366. PMID 23442605. doi:10.1186/1471-2407-13-90.
↑ Cromie, GA; Connelly, JC; Leach, DR (2001). "Recombination at double-strand breaks and DNA ends: conserved mechanisms from phage to humans". Mol Cell. 8 (6): 1163–74. PMID 11779493. doi:10.1016/S1097-2765(01)00419-1.
↑ O'Brien, PJ. (2006). "Catalytic promiscuity and the divergent evolution of DNA repair enzymes". Chem Rev 106 (2): 720–52. PMID 16464022. doi:10.1021/cr040481v.
↑ University of Zurich. (17 de xaneiro de 2013.). ""Great Oxidation Event: More oxygen through multicellularity."". ScienceDaily,.
↑ Maresca, B; Schwartz, JH (2006). "Sudden origins: a general mechanism of evolution based on stress protein concentration and rapid environmental change". Anat Rec B New Anat. 289 (1): 38–46. PMID 16437551. doi:10.1002/ar.b.20089.
↑ DeJong W, Degens H (2011). "The Evolutionary Dynamics of Digital and Nucleotide Codes: A Mutation Protection Perspective" (PDF). Open Evolution Journal, 5: 1-4.
Véxase tamén |
Outros artigos |
- Ciclo celular
- Danos no ADN por causas naturais
- Teoría do envellecemento por danos no ADN
- Replicación do ADN
- Terapia xénica
- Proxeria
- Senescencia
Ligazóns externas |
- 3D structures of some DNA repair enzymes
- Human DNA repair diseases
- DNA Repair
- DNA Damage and DNA Repair
- Segmental Progeria
- DNA-damage repair; the good, the bad, and the ugly
|
|
Categoría:
- Reparación do ADN
(window.RLQ=window.RLQ||[]).push(function()mw.config.set("wgPageParseReport":"limitreport":"cputime":"2.860","walltime":"3.101","ppvisitednodes":"value":8628,"limit":1000000,"ppgeneratednodes":"value":0,"limit":1500000,"postexpandincludesize":"value":302405,"limit":2097152,"templateargumentsize":"value":267,"limit":2097152,"expansiondepth":"value":7,"limit":40,"expensivefunctioncount":"value":3,"limit":500,"unstrip-depth":"value":0,"limit":20,"unstrip-size":"value":234386,"limit":5000000,"entityaccesscount":"value":4,"limit":400,"timingprofile":["100.00% 2382.765 1 -total"," 79.41% 1892.138 1 Modelo:Listaref"," 56.90% 1355.893 123 Modelo:Cita_publicación_periódica"," 10.43% 248.574 1 Modelo:Control_de_autoridades"," 5.65% 134.651 14 Modelo:Cita_libro"," 4.17% 99.415 11 Modelo:Cita_web"," 1.21% 28.950 3 Modelo:Cita_novas"," 0.63% 15.011 1 Modelo:Artigo_de_calidade"," 0.49% 11.733 1 Modelo:Icona_en_título"," 0.13% 3.042 1 Modelo:-"],"scribunto":"limitreport-timeusage":"value":"1.627","limit":"10.000","limitreport-memusage":"value":3303355,"limit":52428800,"cachereport":"origin":"mw1225","timestamp":"20190501164436","ttl":2592000,"transientcontent":false););"@context":"https://schema.org","@type":"Article","name":"Reparaciu00f3n do ADN","url":"https://gl.wikipedia.org/wiki/Reparaci%C3%B3n_do_ADN","sameAs":"http://www.wikidata.org/entity/Q210538","mainEntity":"http://www.wikidata.org/entity/Q210538","author":"@type":"Organization","name":"Contribuidores dos projetos da Wikimedia","publisher":"@type":"Organization","name":"Wikimedia Foundation, Inc.","logo":"@type":"ImageObject","url":"https://www.wikimedia.org/static/images/wmf-hor-googpub.png","datePublished":"2015-03-07T11:11:54Z","dateModified":"2019-05-01T16:44:33Z","image":"https://upload.wikimedia.org/wikipedia/commons/b/b2/Brokechromo.jpg"(window.RLQ=window.RLQ||[]).push(function()mw.config.set("wgBackendResponseTime":152,"wgHostname":"mw1246"););


